- LOGIN
- MemberShip
- 2026-04-24 02:52:26
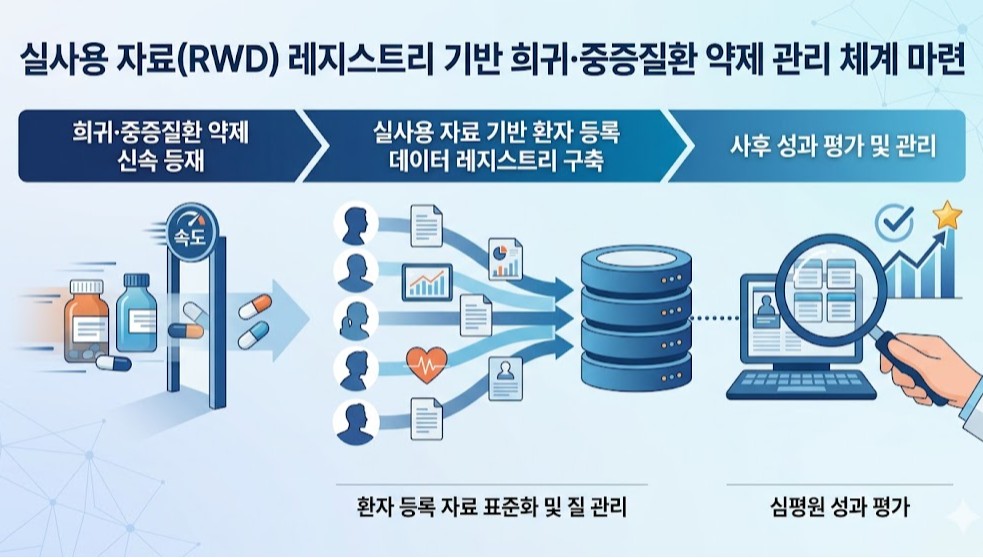
- Policy
- Korea to use RWD for post-listing control of expedited drugs
- by Jung, Heung-Jun Apr 06, 2026 03:51pm
- A plan is being developed to utilize patient registry data, a subset of real-world data (RWD), to strengthen post-listing management of rare and severe disease treatments.This is an extension of the drug pricing system reform announced by the government last month through the National Health Insurance Policy Deliberation Committee. Previously, the government had revealed plans to expedite the listing of treatments for rare and severe diseases and then reevaluate their reimbursement status based on real-world data.According to industry sources on the 6th, HIRA and the Pharmacuetical Performance Assessment Department will conduct a study this year to establish an RWD registry-based management system. A registry refers to patient-level data collected by disease or drug.Although a call for proposals has not yet been issued, the research is scheduled to be completed by the end of this year.Following last year’s study on RWE (Real-World Evidence) guidelines for drug performance assessment, the government is now moving to establish a management system based on RWD registries.This research is significant as it moves beyond the conceptual definition stage towards building an actionable infrastructure. It also serves as a key follow-up measure for implementing the government’s drug pricing system reform.This is because a patient data management system must be established to implement measures such as performance-based reimbursement, adjustments to reimbursement scope, or drug price based on post-marketing evaluation results using real-world data.A HIRA official stated, “Building a registry is essential for managing rare disease treatments. We will conduct policy research on how to actually build such a system. I expect results to be available by the end of the year, following approximately 6 months of research.”The official further explained, “While this can be seen as an extension of the drug pricing system reform, there has long been a consensus that a registry is essential for establishing a management system for rare and severe disease drugs.”However, patient or disease-specific registration data in clinical settings have not yet been standardized, and concerns regarding proper quality control remain unresolved. These issues are expected to be key points of contention during the process of establishing a registry-based management system.An HIRA official stated, “We plan to commission the research soon. We ask researchers to show strong interest, given the high accessibility and utility of the registry data.”
- Company
- Bayer "Pipeline growth accelerate…back on the growth chart"
- by Son, Hyung Min Apr 03, 2026 08:03am
- Bayer is signaling a return to growth trajectory, delivering a series of achievements across its major pipelines. With recent new product approvals and late-stage clinical successes, Bayer's R&D strategic transformation is proving effective.On the 1st (local time), Bayer held its '2026 Pharma Media Day' and announced a medium-to-long-term growth plan based on a science-centered strategy and operational innovation.Stefan Oelrich, Member of the Board of Management of Bayer AG and President of the Pharmaceuticals Division, stated, "Our growth foundation is strengthening based on our focus on strategic priorities and scientific rigor. Through a multimodal pipeline and an AI-based operating model, we aim to recover a growth rate of 4% to 6% starting in 2027 and achieve an operating profit margin of 30% by 2030."Bayer held its '2026 Pharma Media Day'As of 2025, Bayer has secured five approvals, including three new products and two label expansions, and has achieved pipeline milestones by delivering positive results from six Phase 3 clinical trials.Bayer proposed ▲cardiovascular ▲oncology ▲immunology ▲rare diseases ▲women's health as the core pillars that will drive growth over the next decade.In particular, the Factor XIa inhibitor 'Asundexian' secured positive results in reducing the recurrence of ischemic stroke and was designated for Fast Track by the U.S. Food and Drug Administration (FDA), while the prostate cancer treatment 'darolutamide' is undergoing additional clinical trials to expand its treatment lines.Furthermore, 'Kerendia (finerenone)' is now a core growth product, demonstrating clinical utility across multiple Phase 3 studies and a mechanism that simultaneously targets heart and kidney diseases.Bayer also presented precision medicine-based anti-cancer agents and gene·cell therapies as next-generation growth engines.Various drug candidates, such as radiopharmaceutical-based Targeted Alpha Therapy (TAT), WRN inhibitors, and treatments for HER2-mutant lung cancer, are advancing toward clinical trials, while gene and cell therapies targeting Parkinson's disease and heart failure are also under development.In conjunction with this, Bayer is strengthening its integrated therapy-diagnosis strategy by expanding its innovation in diagnostics, including low-dose MRI contrast agents and molecular imaging technology.Bayer's strategy is to increase productivity by integrating AI throughout the entire R&D process.By combining global medical data with AI analysis platforms, Bayer is optimizing the entire process from drug candidate discovery to clinical development, aiming to improve R&D productivity by 40% by 2030.

- Company
- AZ launches triple-combination COPD inhaler Breztri in KOR
- by Son, Hyung Min Apr 03, 2026 08:02am
- Breztri AerosphereAstraZeneca Korea (Country President: Eldana Sauran) announced the launch of ‘Breztri Aerosphere (budesonide, glycopyrronium, formoterol),’ as a maintenance therapy for moderate-to-severe chronic obstructive pulmonary disease (COPD).Breztri Aerosphere is a single-inhaler triple-combination therapy that combines an inhaled corticosteroid (ICS), a long-acting beta-2 agonist (LABA), and a long-acting muscarinic antagonist (LAMA) in a single inhaler. It is indicated as a maintenance treatment for adult COPD patients to control symptoms and reduce exacerbations, and is administered twice daily.The clinical efficacy and safety profile of Breztri Aerosphere have been established through global Phase III trials, including ETHOS and KRONOS.The ETHOS (The Efficacy and Safety of Triple Therapy in Obstructive Lung Disease) study was a 52-week, multicenter, randomized, double-blind Phase III trial involving 8,588 patients aged 40–80 with moderate-to-very severe COPD.Results showed that Breztri Aerosphere reduced the annual rate of moderate or severe COPD exacerbations by approximately 24% compared with LAMA/LABA dual therapy, and by about 13% compared with ICS/LABA, demonstrating statistically significant results.A post-hoc analysis of ETHOS also demonstrated a significant reduction in all-cause mortality in the Breztri treatment group compared with LAMA/LABA.In another pivotal trial, KRONOS, Breztri Aerosphere demonstrated improvements in lung function.The 24-week global Phase III study enrolled 1,902 patients with moderate-to-very severe COPD. At Week 24, Breztri improved lung function by 22 mL compared with LAMA/LABA and by 74 mL compared with ICS/LABA (BFF MDI).COPD is a representative chronic respiratory disease caused by abnormalities in the airways and alveoli, such as bronchitis, bronchiolitis, and emphysema. It is characterized by chronic respiratory symptoms such as shortness of breath and coughing, and is a heterogeneous disease involving persistent and progressive airway obstruction. Compared to the general population, patients with COPD tend to develop various comorbidities at an earlier age. Factors such as smoking, aging, and chronic conditions associated with the disease itself, which include cardiovascular disease, musculoskeletal disorders, and diabetes, further increase disease burden.The 2026 Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines recommend triple therapy with ICS, LAMA, and LABA in patients receiving ICS+LABA who are currently not experiencing an exacerbation but have a high symptom burden, or who are experiencing an exacerbation and have a blood eosinophil count of 100 cells/μL or higher.The guidelines also note that using a single inhaler can improve treatment convenience and adherence compared with multiple inhalers.Ji-young Kim, Executive Vice President of the Respiratory Business Unit at AstraZeneca Korea, stated, “Breztri Aerosphere is a treatment option that has demonstrated reductions in exacerbations and improvements in lung function in global clinical trials, offering a new treatment option for COPD patients in Korea. AstraZeneca Korea will continue to strive to improve the treatment environment and disease management for patients with respiratory diseases.”Breztri Aerosphere is a pMDI (pressurized metered-dose inhaler) designed to uniformly disperse drug particles using Aerosphere delivery technology, ensuring that the medication is delivered throughout both the large and small airways.pMDIs can be used by patients with insufficient inhalation flow rates and offer the advantage of delivering a consistent dose with each actuation. Additionally, as a single inhaler capable of administering three medications simultaneously, it can help improve treatment convenience and medication adherence for patients.

- Company
- "Finished drug firms·API suppliers suffer from high exchange rate"
- by Chon, Seung-Hyun Apr 03, 2026 08:02am
- The pharmaceutical industry is facing significant challenges conducting business amid high exchange rates and the war in the Middle East. As the KRW/USD exchange rate surpasses KRW 1,500, the cost pressure on imported Active Pharmaceutical Ingredients (APIs) is intensifying. Furthermore, with upcoming generic drug price cuts, seeking cheaper imported APIs is becoming increasingly difficult. The high exchange rate and price reductions are also major issues for API manufacturers.According to industry sources on the 2nd, the KRW/USD exchange rate in the Seoul foreign exchange market reached 1,501.5 KRW on the 1st. Compared to KRW 1,352.6 on July 2 of last year, this represents an increase of more than KRW 150 in just eight months.The KRW/USD exchange rate trend (unit: KRW, source: SMB)The exchange rate first surpassed KRW 1,500 on the 24th of last month and briefly stayed in the KRW 1,400 range for two days before surpassing KRW 1,500 again on March 29, continuing to increase. Amid concerns over a prolonged conflict stemming from the high-intensity standoff between the U.S. and Iran, the rate even surged past KRW 1,530 during intraday trading on the 31st, the highest level in 17 years since the global financial crisis in 2009.The decline in the value of the Korean won directly translates to cost-push pressure for pharmaceutical companies. Since these companies are highly dependent on imported APIs, the core raw materials for medicines, the rise in the KRW/USD exchange rate directly increases production costs.In 2024, the self-sufficiency rate for APIs was recorded at 31.4%, calculated using an average exchange rate of KRW 1,367. Self-sufficiency refers to the ratio of domestically produced products within the total market.With 69.6% of APIs used domestically being imported, the reliance on foreign raw materials is absolute. Since US dollars are used even when purchasing APIs from China and India, the largest sources of imports, the impact of the rising exchange rate is unavoidable.Recently, domestic pharmaceutical companies have also been considering switching API suppliers to reduce costs in response to the government's announced price cuts for generic drugs.Under the reformed drug pricing system discussed by the Ministry of Health and Welfare (MOHW) at the Health Insurance Policy Review Committee on the 26th of last month, the price for both off-patent drugs and generics will decrease from 53.55% to 45% of the pre-patent-expiry price of the new drug. Mathematically, this is a 16.0% cut in generic prices.Under the reformed drug pricing system discussed by the Ministry of Health and Welfare (MOHW) at the Health Insurance Policy Review Committee on the 26th of last month, the price for both off-patent drugs and generics will decrease from 53.55% to 45% of the pre-patent-expiry price of the new drug.The price reduction range increases further when "top-tier price requirements," such as conducting bioequivalence (BE) studies and using registered drug substances (DMF), are applied to existing listed generics. Under the reform, the penalty for failing to meet these requirements will expand from 15% to 20%. Since July 2020, requirements have been in place that generics can receive the 53.55% maximum price only if they meet both the direct BE study and DMF criteria. For each unmet requirement, the ceiling price drops by 15%.Applying the new 45% standard and the 20% cut for unmet requirements, generics failing one requirement drop to 36%, and those failing both drop to 28.8%. This means the price for a generic failing one requirement will be 20.9% lower than current levels, while those failing both will see a 25.6% decrease.Due to these price-reduction pressures, pharmaceutical companies are forced to seek cheaper imported products rather than relatively expensive domestic APIs to save costs.The value of API imports from China is already increasing. In 2024, Chinese API imports reached USD 816.32 million, up 110.2% from USD 388.31 million in 2014. In 2014, China was the 6th-largest source of drug imports for Korea, but it rose to 3rd place by 2024.Chinese API Import Amounts by Year (USD 1,000)In 2024, the value of Chinese APIs used in Korea was KRW 1.1159 trillion. Of the KRW 4.4007 trillion in APIs produced in Korea, only KRW 1.43 trillion was used in the domestic market. This indicates that the amount of Korean and Chinese APIs used in the local market is roughly equal. Considering that Chinese APIs are generally cheaper than domestic ones, this suggests that the actual volume of Chinese APIs used by domestic companies overwhelms that of domestic APIs.Under these circumstances, there are concerns that if generic prices drop further, more companies would refrain from using relatively expensive domestic APIs. Both finished drug manufacturers and API suppliers are structured in a way that could lead to simultaneous losses under pressure to reduce drug prices.For API manufacturers, the high exchange rate and price cuts act as major setbacks. Even for domestically produced APIs, starting materials are often imported, so they must worry about rising costs due to exchange rates. As pharmaceutical companies search for cheaper imported alternatives, the concerns of domestic API firms are further compounded.A pharmaceutical industry official stated, "If generic prices fall further, the movement to replace raw materials with cheaper alternatives to save costs will spread. As the dependency on imported APIs increases, domestic API companies find themselves in a position where they must worry about survival."Critics pointed out the ineffectiveness of the government's API preferential pricing policy. The government plans to apply price preferences to listed "essential national medicines" that use domestic APIs. This involves granting a price preference of up to 68% of the pre-patent-expiry price of new drugs for essential medicines made with domestic raw materials.However, pharmaceutical companies complain that the proportion of essential medicines within total drug sales is negligible, and even if prices are raised, there is insufficient incentive to switch to domestic APIs. To receive the preferential designation for domestic raw materials, a company must prove that all raw materials were synthesized at a domestic manufacturing site. Required documents include the ▲API Registration Certificate ▲Common Technical Document (CTD) ▲Manufacturing Instructions and Records.During a parliamentary audit last October, Rep. Baek Jonghean of the People Power Party pointed out, "The fact that not a single pharmaceutical company has applied for the price preference for essential national medicines using domestic raw materials for seven months is proof that the system exists in name only," adding, "Despite industry-wide complaints that the application criteria are too stringent, the MOHW's failure to recognize that the regulations will undermine the policy's ability to foster the domestic API industry."An industry source stated, "Due to the recent aftermath of the war in the Middle East, there are concerns over the supply instability of various raw materials, and with the added burden of costs from the high exchange rate, it is difficult to predict business plans for this year," adding, "In addition to the government's drug price reduction policy, it has become difficult to guarantee the business sustainability of both domestic finished drug and API manufacturers."

- Company
- AstraZeneca Korea appoints Ohad Goldberg as new Country President
- by Son, Hyung Min Apr 03, 2026 08:02am
- Ohad Goldberg, new Country President of AstraZeneca KoreaAstraZeneca Korea announced that it has appointed Ohad Goldberg, Country President of AstraZeneca Israel, as the new Country President of AstraZeneca Korea, effective May 1, 2026.As the new Country President, Goldberg will oversee the company’s business operations in Korea. He plans to expand patient access to AstraZeneca’s medicines and continue to enhance AstraZeneca’s standing as a top-tier partner dedicated to strengthening the domestic life sciences ecosystem for the benefit of Korean patients and society.During his tenure as Country President of AstraZeneca Israel, Goldberg led significant organizational growth and strengthened partnerships across the healthcare ecosystem. With over 20 years of international leadership experience across life sciences, biotech, and ag-tech, he possesses proven capabilities in commercial operations, market access, and external affairs.He has led AstraZeneca Israel’s external partnerships with government, healthcare stakeholders, academia, and innovation platforms. He also served as Chairman of the Board for AION Labs and as a board member of Pharma Israel, representing AstraZeneca in multiple leadership roles.Ohad Goldberg, the new Country Manager of AstraZeneca Korea, said, “It is a great honor to take on this role at a pivotal time for healthcare reform in Korea. Together with our talented team, I look forward to contributing to the advancement of healthcare policy and to creating sustainable, long-term value for Korean patients and society by expanding equitable access to breakthrough medicines.”Goldberg has held senior leadership roles at AstraZeneca, including Global Launch Leader for Respiratory Biologics, Market Access Director, and Respiratory Business Unit Director for Israel, and senior roles in commercial strategy across Europe.
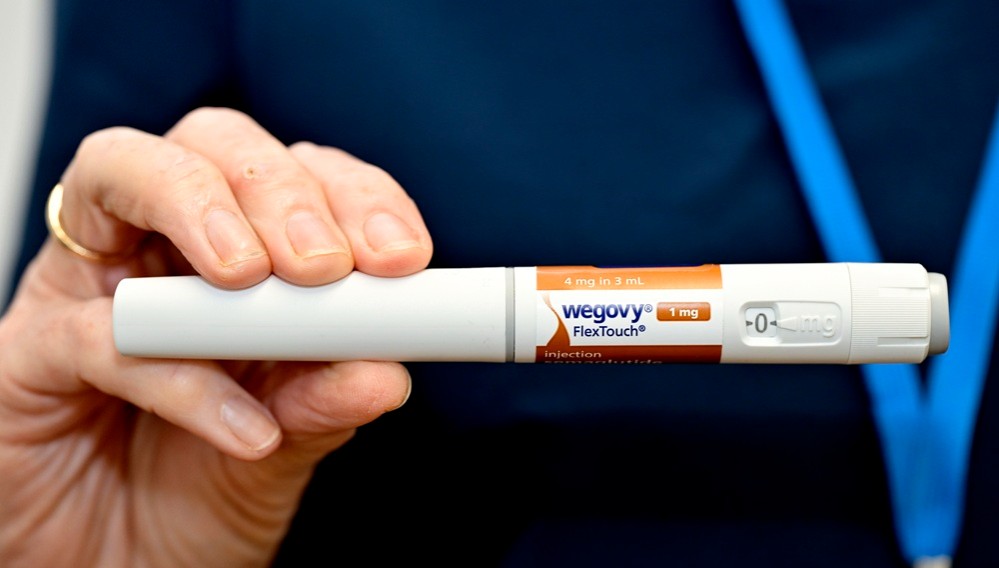
- Company
- Novo Nordisk hits record sales in Korea… led by Wegovy
- by Son, Hyung Min Apr 03, 2026 08:02am
- Novo Nordisk achieved record domestic sales, driven by its obesity treatment 'Wegovy.' As obesity treatments become a primary growth driver, the company’s earnings structure is shifting away from its previous focus on diabetes and hemophilia.According to the Financial Supervisory Service on the 3rd, Novo Nordisk’s sales increased from KRW 308.5 billion in 2024 to KRW 613.6 billion last year, up 85.6% year-on-year. Operating profit rose 77.1% over the same period, from KRW 13.7 billion to KRW 24.2 billion.Novo Nordisk’s performance shows a clear distinction before and after the launch of Wegovy (semaglutide).Prior to the arrival of Wegovy, the company maintained steady growth based on insulin products, hemophilia treatments, and the once-daily obesity drug ‘Saxenda (liraglutide),’ although growth rates were relatively limited.However, its performance surged with the launch of Wegovy in Korea in 2024. With the launch of Wegovy, Novo Nordisk recorded KRW 374.7 billion in sales that year, representing a 62.7% increase from the previous year.According to the market research institution IQVIA, Wegovy generated KRW 467 billion in sales last year, accounting for over 70% of total revenue, and established itself as Novo Nordisk’s core revenue source just one year after its launch. This represents a rare case where a single product drives the growth of a local affiliate.Quarterly trends also highlight rapid growth. Wegovy posted KRW 60.3 billion in Q4 2024, and sales rose to KRW 133.8 billion in Q2 2025, surpassing KRW 100 billion in quarterly sales. It recorded KRW 137 billion and KRW 116.7 billion in Q3 and Q4, respectively, quickly dominating the market.This shift has directly translated into actual market demand.Previously, obesity treatment relied mainly on diet and exercise, with limited use of adjunctive drugs. Injectable treatments like Saxenda existed, but the daily dosing burden and adherence issues constrained market expansion.In contrast, Wegovy demonstrated significant weight loss with once-weekly dosing, greatly improving convenience. Clinical results showing over 15% weight reduction served as a catalyst for shifting obesity treatment from a selective management option to an aggressive treatment option.In Korea, demand surged immediately after launch, leading to supply shortages, with clinics and hospitals reporting a spike in prescription inquiries. This is interpreted not as a temporary trend but as a release of latent demand.As a result, inventories increased sharply. Novo Nordisk’s inventory rose from KRW 80.8 billion in 2024 to KRW 348.2 billion last year, a 331% increase.This reflects the company’s supply expansion strategy to meet surging Wegovy demand. As supply shortages persisted, with repeated sell-outs following the initial launch, the company made a proactive effort to secure inventory and improve its distribution capabilities.In particular, since consistent and stable administration is crucial for obesity treatments, securing inventory is evaluated not merely as an increase in costs but as a key operational indicator supporting revenue growth.Beyond GLP-1 to next-generation mechanisms… Novo Nordisk expands its metabolic pipelineObesity drug ‘Wegovy’Novo Nordisk is globally expanding semaglutide’s indications beyond obesity and diabetes into broader metabolic diseases.Semaglutide has demonstrated reductions in major adverse cardiovascular events (MACE), extending its therapeutic scope beyond diabetes and obesity.By accumulating clinical evidence that encompasses not only diabetes patients but also high-risk cardiovascular groups, it has been clearly demonstrated that GLP-1 agonists can contribute to improving long-term outcomes beyond weight loss.Furthermore, with the addition of chronic kidney disease (CKD) indications, the company is increasing its presence in the renal disease sector previously pioneered by SGLT-2 inhibitors.Recently, semaglutide gained accelerated approval for MASH, further expanding into liver disease.Their mechanistic strengths of weight loss, improvement in insulin resistance, and suppression of inflammation have led to reduced hepatic fat accumulation and improved fibrosis, positioning them as a new alternative in the MASH field, where treatment options have previously been limited.In addition, the company is preparing new drugs with novel mechanisms of action. CagriSema is a combination drug containing 2.4 mg of semaglutide, the active ingredient in Wegovy, and 2.4 mg of the long-acting amylin analog cagrilintide. Cagrilintide mimics the action of amylin, a hormone that naturally suppresses appetite, and is being developed as a once-weekly dose due to its longer duration of action compared to existing treatments.Additionally, the triple agonist ‘UBT251 (GLP-1/GIP/GCG), currently being codeveloped with a Chinese partner, is also emerging as a next-generation growth driver. This is a multi-target drug in the same class as Eli Lilly’s retatrutide. Results of a 24-week Phase II clinical trial recently disclosed in China showed that UBT251 demonstrated a maximum weight loss of 19.7%.If these drugs are introduced to the domestic market, Novo Nordisk’s growth trajectory is expected to accelerate further.

- Policy
- Study to improve ICER threshold underway
- by Jung, Heung-Jun Apr 02, 2026 08:46am
- With the government announcing plans to raise the ICER threshold next year, detailed reform measures are expected to be prepared by October this year.For this, research aimed at ensuring fiscal sustainability while realigning cost-effectiveness evaluation criteria to become more realistic will be conducted over approximately 6 months.On the 31st, the Health Insurance Review and Assessment Service (HIRA) issued a request for proposal for a research project titled “Establishing rational cost-effectiveness evaluation criteria for listing new drugs under the national health insurance system.”This study was launched in response to criticism that the current ICER threshold fails to reflect ▲rising national income and inflation ▲demands for enhanced patient coverage ▲trends in innovative drug development.Through this research, a model for calculating the ICER threshold that incorporates socioeconomic factors such as income and inflation will be developed. A weighting system that takes disease severity, therapeutic benefits, and fiscal impact into account will also be designed.The study will also explore ways to systematize and categorize ICER thresholds and develop models for applying evaluation criteria to reimbursement decisions through pilot implementation.Additionally, the government will review the introduction of a system for periodically adjusting ICER thresholds by referencing major countries. It also plans to gather input from stakeholders, including the pharmaceutical industry, patient groups, and academia.HIRA plans to sign the research service agreement between March and April, conduct an interim review, and complete the study by October. The total budget for the project is KRW 60 million.Through this study, the agency expects to establish rational cost-effectiveness evaluation criteria, thereby improving patient access to treatment and incentivizing new drug development. It also aims to build trust among pharmaceutical companies, patients, and the government by establishing clear standards and procedures.Previously, the Ministry of Health and Welfare announced plans to raise the ICER threshold next year through reforms to the drug pricing system. It decided to introduce a weighting system to allow for the flexible application of ICER values. The ministry plans to conduct policy research this year and implement a rational plan in 2027 based on the results.The government is expected to finalize detailed implementation plans based on the research results, which are due around October, and proceed with full-scale adjustments to the ICER threshold sometime next year.
- InterView
- [Reporter's View] Cut drug prices, expand R&D…SMEs 'exit pressure'
- by Choi Da Eun Apr 02, 2026 08:46am
- The government's recently announced "Drug Pricing System Improvement Plan" appears adequate in terms of its direction. The intent is to improve the market structure, which is currently centered on generics, to provide incentives for pharmaceutical companies to invest in R&D, and to promote self-sufficiency in raw materials. Furthermore, shifting the industrial constitution away from the simple, repetitive manufacturing of off-patent drugs is agreeable.However, the general opinion from the clinical settings is different. There is significant concern that the policy's incentives may actually act as a double burden for many pharmaceutical companies.The core value of this reform is a structure that guarantees relatively higher drug prices for companies with a high R&D investment ratio, in exchange for lower generic drug prices. The higher the ratio of R&D relative to sales, the more a company's generic prices are recognized, which directly correlates to corporate profitability.The problem is that this structure does not operate identically for all companies. While it may serve as a reward for top-tier pharmaceutical companies that have already secured a certain level of R&D capability and capital, it is highly likely to act as a barrier to entry for small and medium-sized enterprises (SMEs).This is because revenue is shrinking as the generic price calculation rate is lowered from 53.55% to 45%, and the timing of "tiered price reductions" for follow-on products applies to the 13th entry. If these tiered price cuts are applied to existing listed products as well, it will be difficult for many SMEs to avoid an immediate drop in cash flow.Of course, the government explains that it will provide incentives through 'Innovative' and 'New Innovative' pharmaceutical company designations. The plan is to guarantee drug prices at a maximum level of 60% for companies with high R&D ratios and apply preferential additions even to companies with a certain level of investment.However, pre-investment is required to meet these standards. A structure where one must increase R&D investment to receive benefits while revenues are decreasing is likely to become a selective incentive that is difficult for companies lacking financial flexibility to access.While the prices of generics, the immediate main source of revenue, are being cut, companies must conversely raise their R&D proportions, which requires pouring tens or hundreds of billions of won, to preserve their lowered profitability. It is a structure where companies are forced into natural extinction in the market if they fail to win the title of a 'New Innovative pharmaceutical company.'A bigger issue is that such a structure could deepen industrial polarization. While top-tier pharmaceutical companies with capital power maximize benefits by expanding R&D investment, SMEs may be pushed out of the market due to limited investment capacity.The government has stated that it will save approximately KRW 2.4 trillion in health insurance costs over the next 11 years through this move, but the price could be the collapse of the ecosystem for smaller pharmaceutical companies that have not yet built up R&D capacity. Some critics argue that this drug pricing reform is essentially a forced restructuring of the domestic pharmaceutical industry.One industry official stated, "While we agree with the direction, the current structure only allows companies with existing capacity to grow further," adding, "Unless realistic support measures that allow SMEs to participate in R&D are implemented as well, the policy effects will inevitably be limited."Pharmaceutical R&D is a long-distance race. It is difficult to evaluate based on short-term results, and the fruits only appear after investing enormous time and effort. In that sense, this drug pricing reform is like placing companies with different levels of physical fitness on the same track from the starting line and demanding they run faster.Furthermore, R&D investment carries the possibility of failure. It is not easy for a company with a weakened profit base to continue bold investments in an area where performance relative to investment is not guaranteed. Consequently, some companies may be driven to focus on securing short-term profits rather than on R&D, or even to downsize their businesses altogether for survival.There is no disagreement with the direction of reducing the overabundance of generics and advancing the industrial structure. However, speed and method are the problem. An approach that induces structural shift solely through uniform price cuts and selective incentives may result in side effects.Unless an environment is established where SMEs can gradually build R&D capabilities while maintaining a minimum investment capacity, this reform is likely to result in industrial contraction rather than promoting innovation. Ultimately, additional incentives must be planned so that policies aimed at lowering drug prices and increasing R&D do not contradict.

- Company
- Imfinzi shifts gastric cancer treatment paradigm
- by Son, Hyung Min Apr 02, 2026 08:46am
- With ‘Imfinzi’ receiving approval as a perioperative treatment for gastric cancer, there are signs that the immunotherapy-plus-chemotherapy strategy, which has long been established as the standard of care overseas, is set to gain full-scale traction in Korea as well.On March 31, AstraZeneca Korea held a press conference at the Four Seasons Hotel in Seoul to share the significance of the expanded indication for Imfinzi (durvalumab) in gastric cancer and its clinical data.On the 23rd, Imfinzi was approved as a perioperative treatment for patients with resectable gastric or gastroesophageal junction adenocarcinoma. The regimen involves combination therapy with FLOT chemotherapy (5-fluorouracil, leucovorin, oxaliplatin, and docetaxel) before surgery, followed by Imfinzi monotherapy as maintenance after surgery.Do-Youn Oh, Professor of Hematology and Oncology at Seoul National University HospitalWith this approval, Imfinzi has become the first immuno-oncology drug approved in Korea for use in the perioperative treatment setting for gastric cancer.Due to advanced screening systems and surgical techniques, the 5-year survival rate for gastric cancer patients in East Asia has been in the 75–80% range with postoperative adjuvant chemotherapy alone.However, approximately 30–40% of stage III patients still experience recurrence, indicating persistent unmet needs.Against this backdrop, perioperative treatment strategies involving chemotherapy before and after surgery have emerged as an alternative.The goal of perioperative therapy is to eliminate micrometastases early and continuously suppress systemic disease thereafter.In the U.S. and Europe, FLOT-based perioperative treatment has already become the standard. The addition of Imfinzi to this regimen has demonstrated significant clinical efficacy, supporting a shift in treatment patterns.Do-Youn Oh, Professor of Hematology and Oncology at Seoul National University Hospital, said, “Perioperative strategies to improve resection rates are already standard overseas. The clinical benefits of combining immunotherapy with chemotherapy are clear.”The Phase III MATTERHORN study was the basis for Imfinzi’s expanded indication. The trial was conducted on patients with stage II-III advanced gastric cancer who were candidates for curative surgery. While stage I gastric cancer has a high cure rate with surgery alone, stages II–III represent locally advanced disease with a higher risk of recurrence.In this study, Imfinzi-based perioperative therapy showed a statistically significant improvement in overall survival (OS).The efficacy of Imfinzi was consistently observed in Asian patients as well.In an Asian subgroup analysis presented at ESMO Asia 2025, the Imfinzi plus FLOT combination demonstrated improvements in event-free survival (EFS), 3-year OS, and pathological complete response (pCR) compared to placebo plus FLOT.At 24 months, the EFS rate was 72.1% in the Imfinzi group versus 64.2% in the placebo group. Median EFS was not reached in either group, suggesting potential widening of the gap with longer follow-up. OS also showed a similar improvement trend to that observed in global studies.The improvement in pCR was particularly notable. In the Asian patient population, the pCR rate in the Imfinzi combination group was 18.9%, more than three times higher than the 5.6% in the placebo group.Safety was also confirmed to be manageable compared to standard FLOT therapy. There were no significant differences between the two groups in Grade 3 or higher adverse events or treatment discontinuation rates, indicating that new safety concerns arising from the addition of an immunotherapy were limited.On the 31st, AstraZeneca Korea held a press conference at the Four Seasons Hotel Seoul to explain changes in treatment strategies following the expansion of Imfinzi’s indication for gastric cancer.Despite surgery remaining the cornerstone of gastric cancer treatment, there is growing recognition that surgery alone may not be sufficient for a cure. The MATTERHORN study suggests that combining immunotherapy and chemotherapy before surgery, followed by surgery and maintenance therapy, can improve long-term outcomes.Professor Oh emphasized, “The proportion of patients completing postoperative Imfinzi adjuvant therapy was around 50%, which exceeded expectations. For patient groups at high risk of micrometastasis, it is important to determine treatment strategies by comprehensively considering various factors such as extensive lymph node involvement, T4 stage, and aggressive histological subtype.”She added, “Clear criteria for determining which patients should undergo surgery first or receive neoadjuvant chemotherapy have not yet been established. Further discussion and accumulation of evidence are necessary to establish treatment strategies tailored to patient characteristics.”

- Policy
- Hair loss drug finasteride, sexual dysfunction-linked suicidal ideation warning
- by Lee, Tak-Sun Apr 02, 2026 08:46am
- AI-generated imageThe labeling for finasteride 1mg tablets, used in the treatment of hair loss, will now include warnings regarding sexual dysfunction side effects.For dutasteride, another hair loss treatment, a new "General Precautions" section will be added to include information on conditions such as depression.The Ministry of Food and Drug Safety (MFDS) recently announced that it has prepared these proposed label changes based on European Medicines Agency (EMA)'s results of a safety information review and has requested feedback from companies by the 10th.According to the revised labeling, a new statement will be added to the warnings section for finasteride 1mg tablets as "Sexual dysfunction that may affect mood changes, including suicidal ideation, has been reported in some patients. Patients should be advised to seek medical consultation if sexual dysfunction occurs. Consideration should be given to whether treatment should be discontinued."While the existing warnings section already mentioned reports of depression, it did not include specific details such as suicidal ideation resulting from sexual dysfunction, as specified in the newly added text.For dutasteride formulations, information regarding 'mood changes and depression' was added to the "General Precautions" section, which is a lower-tier alert than a warning.The newly added information states: "Mood changes (including depressed mood, depression, and rarely, suicidal ideation) have been reported in patients treated with other oral 5-alpha reductase inhibitors. Patients should be advised to seek medical consultation if such symptoms occur."For finasteride 1mg tablets, sexual dysfunction-related adverse events such as decreased libido and erectile dysfunction have already been frequently reported. They are specified in the "Adverse Reactions" section of the labeling. However, details regarding suicidal ideation linked to sexual dysfunction were previously absent.The target products for this label change include 94 items of finasteride 1mg, including the original drug Propecia 1mg.Additionally, for dutasteride, 98 items are subjected to changes, including the original drug Avodart Soft Cap 0.5mg.Meanwhile, in May of last year, the EMA specified suicidal ideation as a new adverse effect in the product information for finasteride. The EMA stated, "Cases have been included where patients complained of suicidal ideation alongside sexual dysfunction such as depression, decreased libido, and erectile dysfunction."The EMA recommended adding a warning statement regarding mood changes and suicidal ideation to dutasteride, a 5-alpha reductase inhibitor of the same class, as a precautionary measure. The EMA stated that, for dutasteride, the evidence does not currently provide a clear causal relationship, unlike that of finasteride.