





-
클래리베이트, 올해의 블록버스터급 신약 후보 발표[데일리팜=노병철 기자] 클래리베이트(Clarivate Plc, NYSE:CLVT)가 2021년 시장에 출시되거나 주요 적응증을 추가할 예정인 의약품들 중 2025년까지 블록버스터급 판매고를 달성할 것으로 예상되는 제품들을 선정한 연례 ‘블록버스터 신약(Drugs to Watch)’ 목록을 발표했다. 신약 개발자들은 코로나19에 대한 업계의 대응 측면에서 전례 없는 성과를 달성하는 것 이상으로 수백만 명의 환자들에게 영향을 미치는 질환에 대한 획기적인 치료법을 발전시키고 있다. 올해의 블록버스터 신약 목록 및 분석 결과는 알츠하이머병과 전립선 암 및 울혈성 심부전 등 만성 진행성 질환임과 동시에 대개 신경쇠약 질환에 해당되는 질병에 대한 치료법에 초점을 맞추고 있다. 바이오 제약기업들은 매우 힘든 한 해였음에도 불구하고 사스-코로나바이러스-2(SARS-CoV-2)를 예방하기 위한 매우 효과적인 백신과 치료제들을 생산했다. 올해의 블록버스터 신약 보고서에는 현재 급부상하고 있는 분야로서 긴급 사용 승인 또는 조건부 승인을 취득한(2021년 2월 10일 기준) 코로나19 백신에 대한 스냅샷 정보 또한 포함되어 있다. 이번 코로나19 팬데믹에 대응하기 위한 조치들, 즉, 더욱 신속한 임상시험의 진행과 투자 급증, 협업 증가, 원격 진료 및 상담 증가 등 업계에서 단행한 일부 조정 사항들을 통해 얻은 교훈들은 즉각적인 위기 대응의 범위를 훨씬 넘어서는 바이오 제약기업의 R&D 및 상업화를 가져올 것으로 전망된다. 클래리베이트는 이미 승인을 받았거나 곧 승인받을 예정인 새로운 의약품 및 생물학적 제제들 중 향후 5년 이내에 10억 달러 이상의 연 매출액을 기록하여 블록버스터급 판매고를 달성할 것으로 예상되는 4개의 치료제를 선정했다. 2021년 블록버스터 신약’ 목록에 포함된 신약은 다음과 같다. 아두카누맙(Aducanumab): 아두카누맙은 현재 전세계 약 5000만명의 환자들에게 영향을 미치고 있는 알츠하이머병에 대한 약전을 구축하기 위한 싸움에서 잠재적인 게임 체인저로 여겨진다. 아두카누맙이 승인될 경우, 이 제품은 알츠하이머병에 대한 최초의 질병완화제제(disease-modifying therapy)가 될 것이며, 환자 치료를 근본적으로 변화시키고, 시장을 변혁시킬 수 있는 획기적인 기회를 가져올 것이다. 또한 승인 이후에 아두카누맙의 수요는 매우 높을 것으로 예상되며, 향후 임상시험에서 임상시험용 약물로 인해 이 치료제를 단념하는 상황도 줄어들 것으로 보인다. 비메키주맙(bimekizumab): 비메키주맙은 전세계 인구의 약 2~3%에 영향을 미치는 질환인 건선 및 기타 다수의 자가면역질환에 시달리고 있는 환자들에게 부작용이 훨씬 적은 치료제가 될 것이다. 비메키주맙은 기존의 치료 옵션들에 비해 점차적으로 개선된 효과를 제공하는 후발 주자이지만, 동급 최고의 효능을 갖추고 있으며 기존 치료제들에 비해 심각한 부작용이 더 적을 것으로 예상된다. 렐루골릭스(Relugolix): 렐루골릭스는 남성들에게 두 번째로 많이 발생하는 악성 종양인 전립선암과 수백만 명의 여성들에게 고통을 주고 있는 자궁 내막증 및 자궁 섬유증을 치료할 수 있는 경구 복용 제제로 동급 최초의 치료제들 중 하나이다. 3가지 적응증에 대한 사용 가능성은 이 제품의 성공 확률을 높여 주고 있다. 렐루골릭스의 경구 제형은 편의성 및 부작용 관리의 용이성 등의 측면에서 주사형 경쟁제품인 성선자극호르몬분비호르몬 작용제(GnRH agonist)에 비해 더 큰 이점을 제공한다. 베리시구아트(Vericiguat): 혁신적인 심부전 치료제인 베리시구아트는 만성 심박출계수 감소 심부전(chronic HFrEF) 고위험군 환자 대상 치료제로서 적용범위가 처음으로 구체적으로 표시된 의약품이다. 베리시구아트의 새로운 작용 기전은 기존 치료법에 대한 추가적인 치료 요법으로 받아들여질 수 있다. 베리시구아트는 고위험 HFrEF 환자들 사이에서 틈새 시장을 찾고, 추가적인 치료 도구로 환영받게 될 것으로 보이며, 해당 환자들의 치료 옵션을 확대할 것으로 예상된다. 클래리베이트의 사고적 리더십, 생명과학 및 보건의료 부문의 글로벌 최고 책임자인 마이크 워드(Mike Ward)는 “우리는 지난 5 년 동안 혁신 의약품에 대한 승인 건수가 10 년 전에 비해 두 배 이상 빠른 속도로 증가하는 것을 보았다. 이러한 성과를 달성하는데 있어 질병의 생물학적 기원에 대한 더 큰 통찰력과 더 나은 표적 치료법을 위한 바이오마커의 정례적 도입을 통한 종양학 연구의 발전, 유전자 및 세포 치료제의 출현, 그리고 의약품 평가를 가속화할 수 있는 프로세스를 도입하기 위한 규제 기관의 노력 등 다양한 동인들이 작용했다”고 말했다. 클래리베이트는 Cortellis Competitive Intelligence™와 Disease Landscape & Forecast™, Drug Timeline & Success Rates™ 및 클래리베이트의 광범위한 인텔리전스 클라우드에 포함된 수천 개의 데이터 포인트에서 얻은 독점적 데이터 및 인사이트를 활용해 2021년 2상 또는 3상 임상시험 단계에 진입한 의약품들 중 올해의 블록버스터 신약 목록을 선정했다. 올해의 블록버스터 신약 목록에 포함된 각 의약품들의 경우 상업적 성공 가능성에 대한 기대가 크며, 일부는 대체 치료제들에 비해 안전성이 높다는 점을 부각시킬 것으로 예상되는 반면 일부 의약품들은 새로운 작용 기전을 강조할 것으로 보인다. 이러한 치료제들은 미래 혁신을 이끌어 낼 뿐만 아니라 인류의 건강을 증진시키고 각 의약품 범주 내에서 치료의 기준을 재정립하며 환자의 생명을 구하거나 삶을 개선할 수 있는 잠재력을 가지고 있다. 클래리베이트는 의약품과 의료 기술 및 의료기기의 라이프사이클 전반에 걸쳐 고객들을 포괄적으로 지원함으로써 인간의 건강을 증진하기 위해 최선을 다하고 있다. 이번 연례 보고서는 클래리베이트와 2020년 클래리베이트가 인수한 DRG(Decision Resources Group)의 결합된 전문지식과 데이터 및 기술이 어떻게 고객들의 데이터 기반 의사결정을 지원하고, 임상 및 상업적 성공에 기여하며, 강력하고 통합된 생명과학 인텔리전스 플랫폼을 강화할 수 있는지를 잘 보여 주고 있다. 2021년 블록버스터 신약 보고서(Drugs to Watch 2021 Report) 전체 내용은 https://discover.clarivate.com/Drugs_to_Watch_2021_download_kr 링크에서 열람 및 다운로드 가능하다.2021-06-30 12:00:07노병철
-
 일동제약 당뇨 신약후보물질 독일 임상1상 돌입[데일리팜=김진구 기자] 일동제약이 개발 중인 제2형 당뇨병치료제 신약 후보물질 'IDG16177'이 독일에서 임상 1상 시험에 돌입한다. & 8203; 일동제약은 지난 28일(현지시간) 독일의 임상승인기관인 연방의약품의료기기관리기관(BfArM)으로부터 신약후보물질 IDG16177에 대한 임상 1상 승인을 받았다고 30일 밝혔다. 일동제약은 이 승인을 바탕으로 곧바로 독일 베를린 현지에서 임상1상에 착수할 계획이다. 2022년까지 임상1상 시험을 완료한다는 목표다. & 8203; IDG16177 임상은 건강한 사람과 당뇨병 환자 총 100여명을 대상으로, IDG16177의 안전성·내약성을 확인하고 유효성 탐색에 초점을 맞춰 계획됐다. & 8203; 건강한 사람으로 구성된 시험군에 IDG16177을 투여한 후 약물 동태, 안전성, 내약성 등을 관찰한다. 이 결과를 바탕으로, 메트포르민 단독요법으로 혈당조절이 충분하지 않은 제2형 당뇨병 환자군에 IDG16177을 메트포르민과 함께 투여하여 당화혈색소(HbA1c) 수치에 미치는 영향을 확인할 계획이다. & 8203; 임상1상 디자인과 투약용량 결정을 위한 PK/PD(약동학/약력학) 시뮬레이션 업무는 일동제약의 연구개발본부와 일동제약그룹의 계열사인 임상약리 컨설팅 전문회사 애임스바이오사이언스와의 협업을 통해 수행됐다. & 8203; IDG16177은 췌장 베타세포의 GPR40(G단백질수용체40)을 활성화해 인슐린 분비를 유도하고 혈당을 조절하는 기전을 가진 GPR40 Agonist 계열의 신약후보물질이다. & 8203; 비임상시험에선 활성·효과·안전성 측면에서 기존 치료제 대비 우위를 확인, 신약으로서의 가능성을 입증했다. 이런 내용은 최근 열린 미국당뇨학회(ADA)에서 공개됐다.2021-06-30 10:33:00김진구
일동제약 당뇨 신약후보물질 독일 임상1상 돌입[데일리팜=김진구 기자] 일동제약이 개발 중인 제2형 당뇨병치료제 신약 후보물질 'IDG16177'이 독일에서 임상 1상 시험에 돌입한다. & 8203; 일동제약은 지난 28일(현지시간) 독일의 임상승인기관인 연방의약품의료기기관리기관(BfArM)으로부터 신약후보물질 IDG16177에 대한 임상 1상 승인을 받았다고 30일 밝혔다. 일동제약은 이 승인을 바탕으로 곧바로 독일 베를린 현지에서 임상1상에 착수할 계획이다. 2022년까지 임상1상 시험을 완료한다는 목표다. & 8203; IDG16177 임상은 건강한 사람과 당뇨병 환자 총 100여명을 대상으로, IDG16177의 안전성·내약성을 확인하고 유효성 탐색에 초점을 맞춰 계획됐다. & 8203; 건강한 사람으로 구성된 시험군에 IDG16177을 투여한 후 약물 동태, 안전성, 내약성 등을 관찰한다. 이 결과를 바탕으로, 메트포르민 단독요법으로 혈당조절이 충분하지 않은 제2형 당뇨병 환자군에 IDG16177을 메트포르민과 함께 투여하여 당화혈색소(HbA1c) 수치에 미치는 영향을 확인할 계획이다. & 8203; 임상1상 디자인과 투약용량 결정을 위한 PK/PD(약동학/약력학) 시뮬레이션 업무는 일동제약의 연구개발본부와 일동제약그룹의 계열사인 임상약리 컨설팅 전문회사 애임스바이오사이언스와의 협업을 통해 수행됐다. & 8203; IDG16177은 췌장 베타세포의 GPR40(G단백질수용체40)을 활성화해 인슐린 분비를 유도하고 혈당을 조절하는 기전을 가진 GPR40 Agonist 계열의 신약후보물질이다. & 8203; 비임상시험에선 활성·효과·안전성 측면에서 기존 치료제 대비 우위를 확인, 신약으로서의 가능성을 입증했다. 이런 내용은 최근 열린 미국당뇨학회(ADA)에서 공개됐다.2021-06-30 10:33:00김진구 -
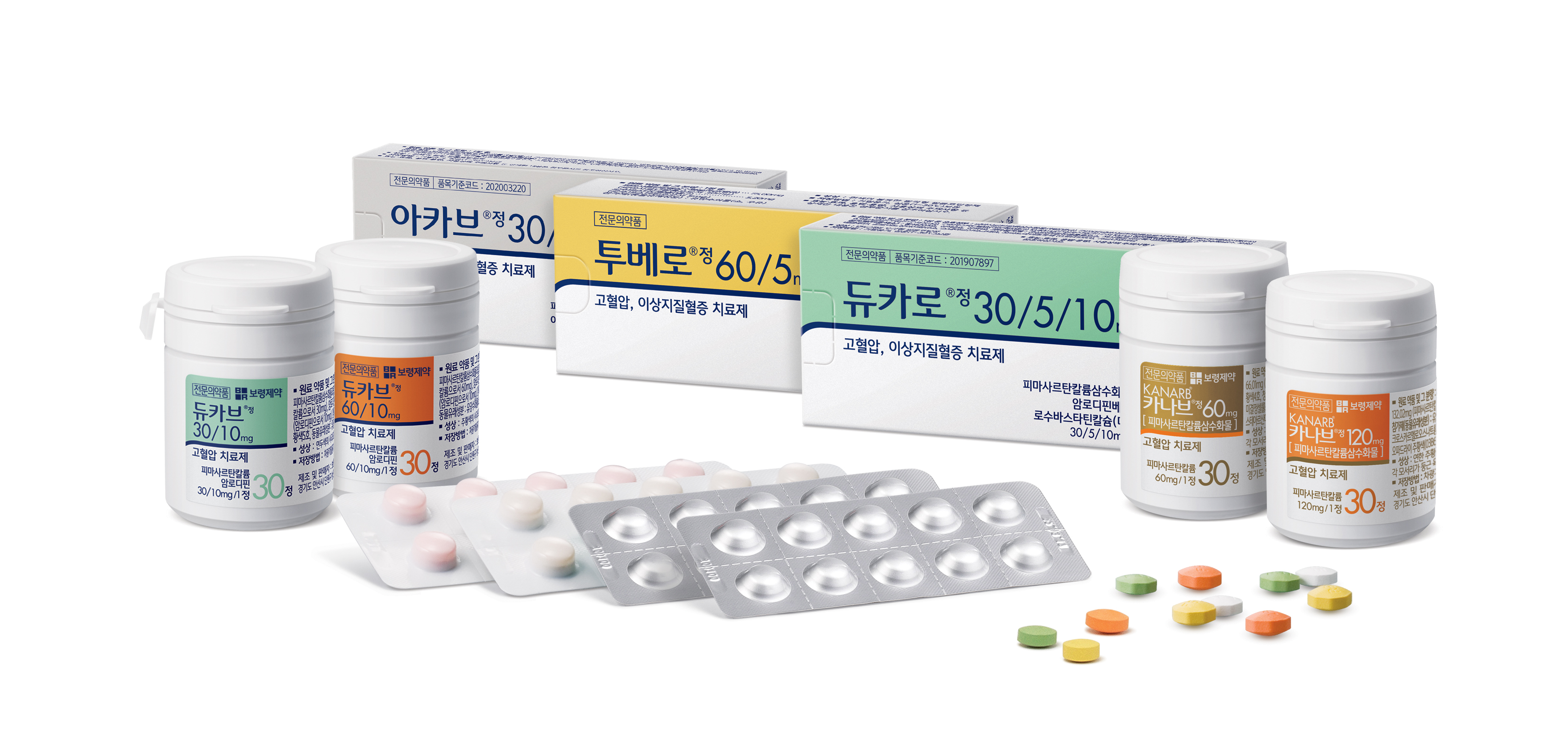 보령제약 "자체 검사결과 카나브서 불순물 미검출"[데일리팜=김진구 기자] 보령제약이 고혈압치료제 카나브의 주성분인 피마사르탄에서 '아지도(AZBT, Azido Methyl Bipheny Tetrazole)' 불순물이 검출되지 않았다고 29일 밝혔다. 보령제약은 최근 식품의약품안전처 권고에 따라 피마사르탄 17개 배지에 대한 시험검사 결과를 진행했다. 앞서 식약처는 해외 불순물 검출 사례를 바탕으로 국내 제약사들에게 6개 ARB 계열 약물에 대한 자체 조사와 시험을 지시한 바 있다. 이르베사르탄·로사르탄·발사르탄에 대해선 불순물 가능성 조사와 시험검사를, 올메사르탄·피마사르탄·칸데사르탄에 대해선 불순물 가능성 조사만 각각 지시했다. 보령제약은 캐나다에서 아지도 불순물 검출 이후 선제적으로 합성공정을 분석한 결과, 아지도 합성 가능성이 없는 것으로 확인됐다고 밝혔다. 여기서 나아가 아지도 불순물 검출을 위한 자체 시험법을 개발해 직접 검사를 실시했다. 17개 배지에 대한 시험검사 결과에서도 아지도 불순물은 검출되지 않았다. 검증 차원에서 진행한 밸리데이션에서도 불순물은 나오지 않았다고 보령제약은 설명했다. 보령제약은 이같은 시험검사 결과를 식약처에 보고했다. 보령제약은 이번 검사를 위해 자체 시험법을 직접 개발했다. 액체크로마토그래프-질량 분석기(LC-MSMS)로 LOQ(정량한계), 검출한계(LOD) 기준을 설정하는 방식이었다. 각 기준은 아지도 불순물 잠정관리기준인 12.5ppm 대비 매우 낮은 극소량으로 설정했다. 매우 적은 불순물이 나오더라도 검출될 수 있도록 정밀하게 설계했다는 설명이다. 이삼수 보령제약 사장은 "최근 NDMA에 이어 아지도까지 불순물에 대한 우려가 커지는 상황에서 자발적·선제적 조치를 통해 안전성을 확인했다"며 "앞으로도 처방의사들과 환자들이 안심하고 처방·복용할 수 있도록 안전한 의약품을 제공하기 위한 투자·역량을 높이겠다"고 말했다.2021-06-29 11:40:06김진구
보령제약 "자체 검사결과 카나브서 불순물 미검출"[데일리팜=김진구 기자] 보령제약이 고혈압치료제 카나브의 주성분인 피마사르탄에서 '아지도(AZBT, Azido Methyl Bipheny Tetrazole)' 불순물이 검출되지 않았다고 29일 밝혔다. 보령제약은 최근 식품의약품안전처 권고에 따라 피마사르탄 17개 배지에 대한 시험검사 결과를 진행했다. 앞서 식약처는 해외 불순물 검출 사례를 바탕으로 국내 제약사들에게 6개 ARB 계열 약물에 대한 자체 조사와 시험을 지시한 바 있다. 이르베사르탄·로사르탄·발사르탄에 대해선 불순물 가능성 조사와 시험검사를, 올메사르탄·피마사르탄·칸데사르탄에 대해선 불순물 가능성 조사만 각각 지시했다. 보령제약은 캐나다에서 아지도 불순물 검출 이후 선제적으로 합성공정을 분석한 결과, 아지도 합성 가능성이 없는 것으로 확인됐다고 밝혔다. 여기서 나아가 아지도 불순물 검출을 위한 자체 시험법을 개발해 직접 검사를 실시했다. 17개 배지에 대한 시험검사 결과에서도 아지도 불순물은 검출되지 않았다. 검증 차원에서 진행한 밸리데이션에서도 불순물은 나오지 않았다고 보령제약은 설명했다. 보령제약은 이같은 시험검사 결과를 식약처에 보고했다. 보령제약은 이번 검사를 위해 자체 시험법을 직접 개발했다. 액체크로마토그래프-질량 분석기(LC-MSMS)로 LOQ(정량한계), 검출한계(LOD) 기준을 설정하는 방식이었다. 각 기준은 아지도 불순물 잠정관리기준인 12.5ppm 대비 매우 낮은 극소량으로 설정했다. 매우 적은 불순물이 나오더라도 검출될 수 있도록 정밀하게 설계했다는 설명이다. 이삼수 보령제약 사장은 "최근 NDMA에 이어 아지도까지 불순물에 대한 우려가 커지는 상황에서 자발적·선제적 조치를 통해 안전성을 확인했다"며 "앞으로도 처방의사들과 환자들이 안심하고 처방·복용할 수 있도록 안전한 의약품을 제공하기 위한 투자·역량을 높이겠다"고 말했다.2021-06-29 11:40:06김진구 -
 반환 신약의 반전...사노피 "한미 신약 심혈관 안전성 확인"[데일리팜=천승현 기자] 사노피가 한미약품에 권리반환한 당뇨신약의 심혈관계와 신장질환 위험성을 줄인다는 대규모 연구 결과를 발표했다. 한미약품은 연구결과를 바탕으로 새로운 기회를 모색하겠다는 구상이다. 29일 한미약품에 따르면 사노피는 미국당뇨병학회(ADA)에서 ‘에페글레나타이드’를 위한 독립세션을 열어 글로벌 대규모 심혈관 임상 3상(AMPLITUDE-O) 결과를 8명의 연구자를 통해 8개의 주제로 나눠 2시간 동안 발표했다. 에페글레나타이드는 지난 2015년 11월 한미약품이 사노피에 기술이전한 GLP-1 계열의 당뇨치료제로, 매일 맞던 주사를 주 1회에서 최장 월 1회까지 연장한 바이오신약이다. 사노피는 에페글레나타이드의 임상3상시험 5건을 진행하다 지난해 9월 최종적으로 권리를 한미약품에 되돌려줬다. 이번에 사노피가 발표한 연구결과는 에페글레나타이드 임상3상시험 5건 중 가장 많은 4000명 이상 환자를 대상으로 진행한 연구다. AMPLITUDE-O 임상 3상은 28개국 344개 지역에서 제2형 당뇨환자나 심혈관 질환 환자 4076명을 대상으로 매주 에페글레나타이드 또는 위약이 투여됐다. 제2형 당뇨환자에서 에페글레나타이드 4mg과 6mg 두 용량 단독 투여 시 심혈관 및 신장질환 발생 위험도가 유의미하게 감소했다. 위약 투여군 대비 에페글레나타이드 투여군에서 주요 심혈관계 질환 발생률은 27%, 신장질환 발생율은 32%로 통계적으로 우월하게 감소했다. 영국 글래스고대학의 나비드 사타(Naveed Sattar) 교수는 “이번 AMPLITUDE-O 임상은 GLP-1 수용체 작용제인 에페글레나타이드가 제2형 당뇨병을 가진 저위험 및 고위험군 환자에서 혈당, 혈압 그리고 체중을 낮추는 가운데 주요 심혈관 및 신장질환의 발생률을 안전하게 감소시켰다”고 평가했다. 한미약품 최고의학책임자 백승재 상무(의학박사)는 “권리 반환을 겪은 에페글레나타이드가 또 다른 혁신을 창출할 수 있는 새로운 기회를 마련했다고 평가하고 싶다”며 “대규모 글로벌 임상 3상을 통해 입증된 에페글레나타이드의 잠재력을 확대하고 구체화하는데 회사 역량을 모으고 있다”고 기대했다. 이번 ADA에서 식이요법과 운동으로 혈당이 조절되지 않는 제2형 당뇨환자 406명을 대상으로 진행한 에페글레나타이드의 또 다른 글로벌 임상 3상(AMPLITUDE-M) 결과도 대표 연구자인 후안 프리아스 박사(Dr. Juan Frias)의 구연 발표로 소개했다. 이중 맹검으로 진행된 이 임상은 56주간 에페글레나타이드를 세 개 용량 투여군(2mg, 4mg, 6mg)으로 나눠 30주차엔 당화혈색소를 1차 평가변수로, 56주차엔 당화혈색소,체중감소,안전성 등을 위약 투여군과 각각 비교했다. 연구 결과 제2형 당뇨환자에게 에페글레나타이드를 투여할 때 우수한 혈당조절 및 체중감소 효과가 확인됐다. 치료효과 또한 장기간 안정적으로 유지됐다. 치료 30주차에 위약 대비 모든 용량에서 당화혈색소가 통계적으로 우월한 개선을 보였다. 이번 발표 직후 국제 학술지인 뉴 잉글랜드 저널 오브 메디슨(New England Journal of Medicine, NEJM)은 에페글레나타이드 관련 논문을 게재했다. 권세창 한미약품 대표이사는 “전세계에서 시판 중인 대사질환 분야 치료제들은 장기 추적 관찰시 심혈관계 질환 유발 가능성을 보이는 경우가 많아 심혈관계 안전성은 약물의 글로벌 경쟁력을 배가하는 중요한 요인이다”며 “대규모 임상에서 에페글레나타이드의 심혈관계 질환 안전성이 입증돼 또 다른 혁신 창출 및 비즈니스 기회를 모색할 수 있게 됐다”고 말했다.2021-06-29 08:32:13천승현
반환 신약의 반전...사노피 "한미 신약 심혈관 안전성 확인"[데일리팜=천승현 기자] 사노피가 한미약품에 권리반환한 당뇨신약의 심혈관계와 신장질환 위험성을 줄인다는 대규모 연구 결과를 발표했다. 한미약품은 연구결과를 바탕으로 새로운 기회를 모색하겠다는 구상이다. 29일 한미약품에 따르면 사노피는 미국당뇨병학회(ADA)에서 ‘에페글레나타이드’를 위한 독립세션을 열어 글로벌 대규모 심혈관 임상 3상(AMPLITUDE-O) 결과를 8명의 연구자를 통해 8개의 주제로 나눠 2시간 동안 발표했다. 에페글레나타이드는 지난 2015년 11월 한미약품이 사노피에 기술이전한 GLP-1 계열의 당뇨치료제로, 매일 맞던 주사를 주 1회에서 최장 월 1회까지 연장한 바이오신약이다. 사노피는 에페글레나타이드의 임상3상시험 5건을 진행하다 지난해 9월 최종적으로 권리를 한미약품에 되돌려줬다. 이번에 사노피가 발표한 연구결과는 에페글레나타이드 임상3상시험 5건 중 가장 많은 4000명 이상 환자를 대상으로 진행한 연구다. AMPLITUDE-O 임상 3상은 28개국 344개 지역에서 제2형 당뇨환자나 심혈관 질환 환자 4076명을 대상으로 매주 에페글레나타이드 또는 위약이 투여됐다. 제2형 당뇨환자에서 에페글레나타이드 4mg과 6mg 두 용량 단독 투여 시 심혈관 및 신장질환 발생 위험도가 유의미하게 감소했다. 위약 투여군 대비 에페글레나타이드 투여군에서 주요 심혈관계 질환 발생률은 27%, 신장질환 발생율은 32%로 통계적으로 우월하게 감소했다. 영국 글래스고대학의 나비드 사타(Naveed Sattar) 교수는 “이번 AMPLITUDE-O 임상은 GLP-1 수용체 작용제인 에페글레나타이드가 제2형 당뇨병을 가진 저위험 및 고위험군 환자에서 혈당, 혈압 그리고 체중을 낮추는 가운데 주요 심혈관 및 신장질환의 발생률을 안전하게 감소시켰다”고 평가했다. 한미약품 최고의학책임자 백승재 상무(의학박사)는 “권리 반환을 겪은 에페글레나타이드가 또 다른 혁신을 창출할 수 있는 새로운 기회를 마련했다고 평가하고 싶다”며 “대규모 글로벌 임상 3상을 통해 입증된 에페글레나타이드의 잠재력을 확대하고 구체화하는데 회사 역량을 모으고 있다”고 기대했다. 이번 ADA에서 식이요법과 운동으로 혈당이 조절되지 않는 제2형 당뇨환자 406명을 대상으로 진행한 에페글레나타이드의 또 다른 글로벌 임상 3상(AMPLITUDE-M) 결과도 대표 연구자인 후안 프리아스 박사(Dr. Juan Frias)의 구연 발표로 소개했다. 이중 맹검으로 진행된 이 임상은 56주간 에페글레나타이드를 세 개 용량 투여군(2mg, 4mg, 6mg)으로 나눠 30주차엔 당화혈색소를 1차 평가변수로, 56주차엔 당화혈색소,체중감소,안전성 등을 위약 투여군과 각각 비교했다. 연구 결과 제2형 당뇨환자에게 에페글레나타이드를 투여할 때 우수한 혈당조절 및 체중감소 효과가 확인됐다. 치료효과 또한 장기간 안정적으로 유지됐다. 치료 30주차에 위약 대비 모든 용량에서 당화혈색소가 통계적으로 우월한 개선을 보였다. 이번 발표 직후 국제 학술지인 뉴 잉글랜드 저널 오브 메디슨(New England Journal of Medicine, NEJM)은 에페글레나타이드 관련 논문을 게재했다. 권세창 한미약품 대표이사는 “전세계에서 시판 중인 대사질환 분야 치료제들은 장기 추적 관찰시 심혈관계 질환 유발 가능성을 보이는 경우가 많아 심혈관계 안전성은 약물의 글로벌 경쟁력을 배가하는 중요한 요인이다”며 “대규모 임상에서 에페글레나타이드의 심혈관계 질환 안전성이 입증돼 또 다른 혁신 창출 및 비즈니스 기회를 모색할 수 있게 됐다”고 말했다.2021-06-29 08:32:13천승현 -
 K-코로나백신 첫 3상 초읽기…국내사들, 개발 잰걸음[데일리팜=김진구 기자] 코로나 백신 개발에 뛰어든 국내제약사들이 개발 속도를 높이는 모습이다. 이르면 내년 상반기 '국산 코로나 백신'이 탄생할 가능성이 점쳐진다. SK바이오사이언스는 국내제약사 중에는 처음으로 코로나 백신 관련 임상3상 시험계획서를 제출했다. 내달 중으로 임상3상에 돌입한다는 계획이다. 제넥신은 대규모 글로벌 임상3상을 위해 1200억원 규모의 자금 유치에 나선 것으로 전해진다. 셀리드와 유바이오로직스는 나란히 임상2상을 진행 중이다. 두 회사는 올해 안에 임상2상 결과를 확보한다는 방침이다. ◆SK바사 "내달 최종후보 선정…4천명 규모 국내외 3상 진행" 29일 제약업계에 따르면 현재 국내에선 5개 제약사가 6개 후보물질로 코로나 백신을 개발 중이다. SK바이오사이언스 2종, 제넥신·셀리드·유바이오로직스·진원생명과학 각 1종의 후보물질을 보유하고 있다. 이들은 대부분 내년 상반기 상용화를 목표로 하고 있다. 가장 관심을 모으는 것은 SK바이오사이언스다. 2종의 후보물질, GBP510과 NBP2001의 임상3상 진입이 가시권에 들어왔다. 지난 28일엔 GBP510의 임상3상 시험계획서를 식품의약품안전처에 제출했다. 국내제약사 중 코로나 백신 임상3상 신청은 SK바이오사이언스가 처음이다. 곧이어 NBP2001의 임상3상 시험계획서도 제출할 것으로 보인다. SK바이오사이언스는 GBP510과 NBP2001의 임상1상 결과를 바탕으로 둘 중 하나를 최종 후보로 선정, 임상3상에 본격 돌입한다는 방침이다. 실무적으로 식약처와 최종후보 선정을 위한 세부협의를 이어가고 있다. 임상3상은 국내와 해외에서 동시 진행된다. 총 규모는 4000여명이 될 것으로 예상된다. 국내에선 14개 의료기관이 선정됐다. 해외임상은 유럽과 동남아 일부 국가에서 진행할 계획이다. SK바이오사이언스 백신은 합성항원 방식으로 개발된다. 글로벌 제약사 백신 가운데선 노바백스 백신과 방식이 같다. 식약처는 같은 방식의 백신과의 비교임상(3상)을 허용한 상태다. 접종 후 4주 시점에서 대조약(기존 허가백신)과 중화항체를 비교해 열등하지 않거나 우월하다는 점을 입증하면 된다. 기존 방식으론 최소 3만명 이상이던 임상규모가 4000명 수준으로 감소한다. 기업의 부담이 줄어듦과 동시에 개발속도가 빨라진다. SK바이오사이언스가 목표로 한 상용화 시점은 내년 상반기다. SK바이오사이언스 관계자는 "NBP2001의 임상1상도 조만간 마무리된다. 7월 중 두 후보물질 중 최종후보를 선정하고 국내외에서 임상3상에 진입한다는 계획"이라며 "내년 상반기 상용화가 목표"라고 말했다. ◆제넥신, 대규모 3상 준비 중…"동남아서 5천명 확보" 국내에서 가장 먼저 코로나 백신 개발에 뛰어든 제넥신도 내년 상반기를 출시 목표시점으로 잡고 있다. 제넥신은 GX-19N이란 이름으로 코로나 백신을 개발 중이다. 임상1상이 마무리됐다. 이달 초 발표된 임상1상 결과에선, 임상참가자 21명 가운데 81%인 17명에서 스파이크단백질 결합 항체가 4배 이상 증가한 것으로 나타났다. 회사는 중화항체도 유의하게 증가했다고 설명했다. 피험자 10명에서 약물 이상반응이 나타났지만 모두 경미한 수준이었다. 이어 진행한 임상2a상도 150명 규모의 피험자 투약이 마무리된 상태다. 이를 토대로 올 하반기에는 임상3상에 착수한다는 것이 회사의 목표다. 이어 내년 상반기엔 긴급사용승인을 받겠다는 계획이다. 관건은 임상규모다. DNA백신 플랫폼을 사용하고 있어, 다른 백신과 달리 마땅한 비교군이 없다. 3만명 이상의 대규모 임상시험이 불가피하다는 설명이다. 이를 위해 제넥신은 해외로 눈을 돌렸다. 인도네시아에서 임상시험 계약 체결을 통해 5000명을 확보했다. 여기에 터키와 아르헨티나 등에서 임상3상을 동시 진행하는 방안을 검토 중이다. 또 1200억원 규모의 자금을 유치하기 위해 전환사채 발행을 검토 중인 것으로도 알려졌다. 대규모 임상3상에 들어가는 비용을 조달하기 위한 목적이라는 해석이 나왔다. ◆임상 변경한 셀리드…"유효성 문제 아닌 대량생산 목적" 셀리드는 이달 초 임상1/2a상 시험계획의 변경을 신청했다. 기존에 개발하던 'AdCLD-CoV19'에 더해 신규 후보물질인 'AdCLD-CoV19-1'의 1상과 2a상을 진행하는 것이 골자다. 셀리드는 임상 변경 이유에 대해 "기존 후보물질에 안전성·유효성 등 문제가 있는 건 아니다"라며 "생산성을 높이기 위해 후보물질을 추가한 것"이라고 설명했다. 기존 후보물질은 생산성이 다소 떨어지는 것으로 전해진다. 이를 극복하기 위해 기본이 되는 바이러스벡터의 종류를 조금 바꿔 대량생산이 가능한 후보물질의 개발에 동시에 나선다는 게 셀리드의 계획이다. 기존 후보물질의 경우 임상2a상 피험자 투약까지 마무리됐다. 현재는 결과를 분석 중이다. 이어 7월 중 임상2b상에, 9월 중 임상3상에 진입하겠다는 목표를 세웠다. 셀리드의 임상3상은 아스트라제네카 혹은 얀센 백신과의 비교임상으로 진행될 것으로 예상된다. 이미 임상3상을 위한 시험용의약품 생산까지 돌입한 것으로 전해진다. 셀리드는 계획대로 임상이 순조롭게 진행되면 내년 상반기엔 상용화가 가능할 것으로 내다보고 있다. ◆유바이오 "10월 2상 결과 발표"…진원생명 "내달 2a상 착수" 유바이오로직스는 '유코백19'라는 이름으로 백신을 개발 중이다. 현재 임상2상에 돌입했으며, 당장 다음 주부터 피험자 투약을 시작한다는 계획이다. 유바이오로직스는 임상2상 참여자를 대부분 모집한 상태로, 순조롭게 진행될 것으로 예상하고 있다. 유바이오로직스 관계자는 "9월 초면 투약이 마무리될 것으로 예상한다. 데이터 수집·분석에 걸리는 시간을 감안하면, 10월에는 대략적인 결과를 확인할 수 있을 것"이라고 말했다. 유바이오로직스는 임상2상이 마무리 되는대로 3상에 나설 계획이다. 임상3상은 노바백스 백신과의 비교임상이 유력하다. 이를 통해 내년 상반기 상용화를 목표로 하고 있다. 진원생명과학은 'GLS-5130'이란 이름으로 백신을 개발하고 있다. 임상1상 투약이 마무리됐다. 7월 중에는 임상2a상에 착수한다는 계획이다. 이어 연내 임상3상을 진행하고, 긴급사용승인을 통해 내년 상반기 백신 출시를 목표로 하고 있다. 제넥신과 마찬가지로 임상3상이 관건이 될 것으로 보인다. 진원생명과학은 제넥신과 같은 DNA백신 플랫폼을 사용하고 있다. 대규모 임상3상이 불가피한 상황에서 진원생명과학은 한국과 인도 등에서 임상을 동시 진행하겠다는 계획을 세웠다.2021-06-29 06:19:18김진구
K-코로나백신 첫 3상 초읽기…국내사들, 개발 잰걸음[데일리팜=김진구 기자] 코로나 백신 개발에 뛰어든 국내제약사들이 개발 속도를 높이는 모습이다. 이르면 내년 상반기 '국산 코로나 백신'이 탄생할 가능성이 점쳐진다. SK바이오사이언스는 국내제약사 중에는 처음으로 코로나 백신 관련 임상3상 시험계획서를 제출했다. 내달 중으로 임상3상에 돌입한다는 계획이다. 제넥신은 대규모 글로벌 임상3상을 위해 1200억원 규모의 자금 유치에 나선 것으로 전해진다. 셀리드와 유바이오로직스는 나란히 임상2상을 진행 중이다. 두 회사는 올해 안에 임상2상 결과를 확보한다는 방침이다. ◆SK바사 "내달 최종후보 선정…4천명 규모 국내외 3상 진행" 29일 제약업계에 따르면 현재 국내에선 5개 제약사가 6개 후보물질로 코로나 백신을 개발 중이다. SK바이오사이언스 2종, 제넥신·셀리드·유바이오로직스·진원생명과학 각 1종의 후보물질을 보유하고 있다. 이들은 대부분 내년 상반기 상용화를 목표로 하고 있다. 가장 관심을 모으는 것은 SK바이오사이언스다. 2종의 후보물질, GBP510과 NBP2001의 임상3상 진입이 가시권에 들어왔다. 지난 28일엔 GBP510의 임상3상 시험계획서를 식품의약품안전처에 제출했다. 국내제약사 중 코로나 백신 임상3상 신청은 SK바이오사이언스가 처음이다. 곧이어 NBP2001의 임상3상 시험계획서도 제출할 것으로 보인다. SK바이오사이언스는 GBP510과 NBP2001의 임상1상 결과를 바탕으로 둘 중 하나를 최종 후보로 선정, 임상3상에 본격 돌입한다는 방침이다. 실무적으로 식약처와 최종후보 선정을 위한 세부협의를 이어가고 있다. 임상3상은 국내와 해외에서 동시 진행된다. 총 규모는 4000여명이 될 것으로 예상된다. 국내에선 14개 의료기관이 선정됐다. 해외임상은 유럽과 동남아 일부 국가에서 진행할 계획이다. SK바이오사이언스 백신은 합성항원 방식으로 개발된다. 글로벌 제약사 백신 가운데선 노바백스 백신과 방식이 같다. 식약처는 같은 방식의 백신과의 비교임상(3상)을 허용한 상태다. 접종 후 4주 시점에서 대조약(기존 허가백신)과 중화항체를 비교해 열등하지 않거나 우월하다는 점을 입증하면 된다. 기존 방식으론 최소 3만명 이상이던 임상규모가 4000명 수준으로 감소한다. 기업의 부담이 줄어듦과 동시에 개발속도가 빨라진다. SK바이오사이언스가 목표로 한 상용화 시점은 내년 상반기다. SK바이오사이언스 관계자는 "NBP2001의 임상1상도 조만간 마무리된다. 7월 중 두 후보물질 중 최종후보를 선정하고 국내외에서 임상3상에 진입한다는 계획"이라며 "내년 상반기 상용화가 목표"라고 말했다. ◆제넥신, 대규모 3상 준비 중…"동남아서 5천명 확보" 국내에서 가장 먼저 코로나 백신 개발에 뛰어든 제넥신도 내년 상반기를 출시 목표시점으로 잡고 있다. 제넥신은 GX-19N이란 이름으로 코로나 백신을 개발 중이다. 임상1상이 마무리됐다. 이달 초 발표된 임상1상 결과에선, 임상참가자 21명 가운데 81%인 17명에서 스파이크단백질 결합 항체가 4배 이상 증가한 것으로 나타났다. 회사는 중화항체도 유의하게 증가했다고 설명했다. 피험자 10명에서 약물 이상반응이 나타났지만 모두 경미한 수준이었다. 이어 진행한 임상2a상도 150명 규모의 피험자 투약이 마무리된 상태다. 이를 토대로 올 하반기에는 임상3상에 착수한다는 것이 회사의 목표다. 이어 내년 상반기엔 긴급사용승인을 받겠다는 계획이다. 관건은 임상규모다. DNA백신 플랫폼을 사용하고 있어, 다른 백신과 달리 마땅한 비교군이 없다. 3만명 이상의 대규모 임상시험이 불가피하다는 설명이다. 이를 위해 제넥신은 해외로 눈을 돌렸다. 인도네시아에서 임상시험 계약 체결을 통해 5000명을 확보했다. 여기에 터키와 아르헨티나 등에서 임상3상을 동시 진행하는 방안을 검토 중이다. 또 1200억원 규모의 자금을 유치하기 위해 전환사채 발행을 검토 중인 것으로도 알려졌다. 대규모 임상3상에 들어가는 비용을 조달하기 위한 목적이라는 해석이 나왔다. ◆임상 변경한 셀리드…"유효성 문제 아닌 대량생산 목적" 셀리드는 이달 초 임상1/2a상 시험계획의 변경을 신청했다. 기존에 개발하던 'AdCLD-CoV19'에 더해 신규 후보물질인 'AdCLD-CoV19-1'의 1상과 2a상을 진행하는 것이 골자다. 셀리드는 임상 변경 이유에 대해 "기존 후보물질에 안전성·유효성 등 문제가 있는 건 아니다"라며 "생산성을 높이기 위해 후보물질을 추가한 것"이라고 설명했다. 기존 후보물질은 생산성이 다소 떨어지는 것으로 전해진다. 이를 극복하기 위해 기본이 되는 바이러스벡터의 종류를 조금 바꿔 대량생산이 가능한 후보물질의 개발에 동시에 나선다는 게 셀리드의 계획이다. 기존 후보물질의 경우 임상2a상 피험자 투약까지 마무리됐다. 현재는 결과를 분석 중이다. 이어 7월 중 임상2b상에, 9월 중 임상3상에 진입하겠다는 목표를 세웠다. 셀리드의 임상3상은 아스트라제네카 혹은 얀센 백신과의 비교임상으로 진행될 것으로 예상된다. 이미 임상3상을 위한 시험용의약품 생산까지 돌입한 것으로 전해진다. 셀리드는 계획대로 임상이 순조롭게 진행되면 내년 상반기엔 상용화가 가능할 것으로 내다보고 있다. ◆유바이오 "10월 2상 결과 발표"…진원생명 "내달 2a상 착수" 유바이오로직스는 '유코백19'라는 이름으로 백신을 개발 중이다. 현재 임상2상에 돌입했으며, 당장 다음 주부터 피험자 투약을 시작한다는 계획이다. 유바이오로직스는 임상2상 참여자를 대부분 모집한 상태로, 순조롭게 진행될 것으로 예상하고 있다. 유바이오로직스 관계자는 "9월 초면 투약이 마무리될 것으로 예상한다. 데이터 수집·분석에 걸리는 시간을 감안하면, 10월에는 대략적인 결과를 확인할 수 있을 것"이라고 말했다. 유바이오로직스는 임상2상이 마무리 되는대로 3상에 나설 계획이다. 임상3상은 노바백스 백신과의 비교임상이 유력하다. 이를 통해 내년 상반기 상용화를 목표로 하고 있다. 진원생명과학은 'GLS-5130'이란 이름으로 백신을 개발하고 있다. 임상1상 투약이 마무리됐다. 7월 중에는 임상2a상에 착수한다는 계획이다. 이어 연내 임상3상을 진행하고, 긴급사용승인을 통해 내년 상반기 백신 출시를 목표로 하고 있다. 제넥신과 마찬가지로 임상3상이 관건이 될 것으로 보인다. 진원생명과학은 제넥신과 같은 DNA백신 플랫폼을 사용하고 있다. 대규모 임상3상이 불가피한 상황에서 진원생명과학은 한국과 인도 등에서 임상을 동시 진행하겠다는 계획을 세웠다.2021-06-29 06:19:18김진구 -
 SK바이오사이언스, 코로나백신 임상3상계획서 제출[데일리팜=김진구 기자] SK바이오사이언스는 28일 코로나19 백신으로 개발 중인 'GBP510'의 임상3상 시험계획서를 식품의약품안전처에 제출했다고 밝혔다. 식약처가 SK바이오사이언스의 임상계획을 승인하면 GBP510는 국내개발 백신 중 처음으로 임상3상에 돌입하는 백신이 된다. SK바이오사이언스는 임상시험계획서 제출에 이어 식약처와 세부협의를 이어간다는 방침이다. 개발속도를 높이기 위한 전략이다. 특히 또 다른 후보물질인 NBP2001의 임상결과를 확인하면서 최종 후보를 선정해 본격적인 임상3상으로 이어갈 계획이다. 이를 위해 SK바이오사이언스는 식약처와 세부 협의를 진행키로 했다. 국내 임상3상 개시시점과 맞춰 글로벌 임상도 동시에 진행할 방침이다. IVI(국제백신연구소)와 함께 유럽·동남아에서 임상3상에 돌입한다는 것이 SK바이오사이언스의 계획이다. 임상3상은 국내 14개 기관을 포함한 다국가 기관에서 건강한 성인 4000여명을 대상으로 진행된다. 중화항체가 등 면역원성 지표와 부작용을 확인하는 내용이다. 임상이 순조롭게 마무리되면 내년 상반기 상용화가 가능할 것으로 SK바이오사이언스는 내다보고 있다. 앞서 SK바이오사이언스는 빌&멜린다게이츠재단·CEPI(전염병대비혁신연합) 등의 지원을 받아 미국 워싱턴대 항원디자인연구소(Institute for Protein Design, IPD)와 공동 개발한 코로나19 백신을 개발해왔다. CEPI는 지난해 GBP510을 'Wave2 프로젝트'로 선정한 바 있다. Wave2는 차세대 코로나 백신 개발을 목표로 하는 프로젝트다. 보관방법·접종횟수·생산성·면역반응 등에서 기존 백신보다 보편적이고 경제적인 백신을 개발하는 것이 프로젝트의 목표다. SK바이오사이언스가 임상3상을 통해 유효성·안전성을 입증하고 상용화까지 성공하면 '코백스 퍼실리티'(COVAX facility)를 통해 전 세계에 수억 회 접종분이 공급된다.2021-06-28 16:52:32김진구
SK바이오사이언스, 코로나백신 임상3상계획서 제출[데일리팜=김진구 기자] SK바이오사이언스는 28일 코로나19 백신으로 개발 중인 'GBP510'의 임상3상 시험계획서를 식품의약품안전처에 제출했다고 밝혔다. 식약처가 SK바이오사이언스의 임상계획을 승인하면 GBP510는 국내개발 백신 중 처음으로 임상3상에 돌입하는 백신이 된다. SK바이오사이언스는 임상시험계획서 제출에 이어 식약처와 세부협의를 이어간다는 방침이다. 개발속도를 높이기 위한 전략이다. 특히 또 다른 후보물질인 NBP2001의 임상결과를 확인하면서 최종 후보를 선정해 본격적인 임상3상으로 이어갈 계획이다. 이를 위해 SK바이오사이언스는 식약처와 세부 협의를 진행키로 했다. 국내 임상3상 개시시점과 맞춰 글로벌 임상도 동시에 진행할 방침이다. IVI(국제백신연구소)와 함께 유럽·동남아에서 임상3상에 돌입한다는 것이 SK바이오사이언스의 계획이다. 임상3상은 국내 14개 기관을 포함한 다국가 기관에서 건강한 성인 4000여명을 대상으로 진행된다. 중화항체가 등 면역원성 지표와 부작용을 확인하는 내용이다. 임상이 순조롭게 마무리되면 내년 상반기 상용화가 가능할 것으로 SK바이오사이언스는 내다보고 있다. 앞서 SK바이오사이언스는 빌&멜린다게이츠재단·CEPI(전염병대비혁신연합) 등의 지원을 받아 미국 워싱턴대 항원디자인연구소(Institute for Protein Design, IPD)와 공동 개발한 코로나19 백신을 개발해왔다. CEPI는 지난해 GBP510을 'Wave2 프로젝트'로 선정한 바 있다. Wave2는 차세대 코로나 백신 개발을 목표로 하는 프로젝트다. 보관방법·접종횟수·생산성·면역반응 등에서 기존 백신보다 보편적이고 경제적인 백신을 개발하는 것이 프로젝트의 목표다. SK바이오사이언스가 임상3상을 통해 유효성·안전성을 입증하고 상용화까지 성공하면 '코백스 퍼실리티'(COVAX facility)를 통해 전 세계에 수억 회 접종분이 공급된다.2021-06-28 16:52:32김진구 -
 브릿지바이오 "베링거 반환 신약, 추가 독성실험 추진"[데일리팜=안경진 기자] 브릿지바이오테라퓨틱스는 특발성 폐섬유증 치료후보물질 'BBT-877' 개발과 관련해 미국식품의약국(FDA)으로부터 서면 회신을 수령했다고 28일 공시했다. 'BBT-877'의 추가 실험을 거쳐 임상2상 설계를 공고히 하라는 FDA의 권고사항을 받아들여 연말까지 추가 실험을 진행하고 관련 데이터를 확보하겠다는 방침이다. 'BBT-877'은 브릿지바이오가 지난 2019년 7월 베링거인겔하임에 기술이전했다가 작년 11월 권리를 돌려받은 특발성 폐섬유증 신약후보물질이다. 다양한 세포종에서 섬유화를 관할하는 오토택신 효소를 저해하는 기전으로 작용한다. 브릿지바이오가 원개발사인 레고켐바이오로부터 'BBT-877'를 도입한 다음 1상임상 단계에서 기술이전 계약을 체결했는데, 임상 1상 당시 병행한 비임상 실험에서 잠재적 독성 우려가 제기되면서 최종적으로 권리 반환이 결정됐다. 브릿지바이오는 FDA와 지난 3월 'BBT-877' 개발 관련 회의를 진행한지 3개월 여만에 서면으로 FDA 입장을 전달받은 상황이다. 브릿지바이오에 따르면 FDA는 'BBT-877' 관련 In vivo GLP 혜성 분석과 전자현미경을 통한 세포사멸 분석 등의 비임상 데이터를 추가로 제출하라고 권고했다. 브릿지바이오는 자체 및 외부 실험을 통해 약물의 직접적 DNA 손상이 아닌, 고농도 약물 처리로 인한 세포사멸 기전이 'BBT-877'의 잠재적 독성 우려를 발생시킨 원인이라고 보고 있다. 이같은 입장을 FDA에 소명하고, 독성반응이 위양성이라는 사실을 재확인하기 위해 추가 실험을 진행하기로 협의한 것이다. 브릿지바이오는 이번에 수령한 FDA 권고에 따라 ▲생체 내 실험을 통한 혜성 분석(in vivo Comet assay) ▲닌테다닙 및 피르페니돈 등 특발성 폐섬유증 환자에서의 기존 표준 치료제와의 약물상호작용(DDI) 시험에 곧바로 착수하겠다고 밝혔다. 올해 연말까지 두 데이터를 추가로 확보하고, FDA와 추가적인 상의 절차를 거쳐 임상 2상의 최종 설계를 구체화한다는 계획이다. 내년 상반기에는 임상 2상의 시험계획신청(CTA)을 완료하고 임상 개시가 가능할 것으로 내다봤다. 특발성 폐섬유증 외에 진행성 표현형을 나타내는 만성 섬유성 간질성폐질환(PF-ILD) 및 다양한 암종을 타깃하는 적응증 확대 연구도 연내 구체화한다는 방침이다. 올해 초 경쟁사인 갈라파고스가 동일 계열의 신약후보물질 ‘GLPG1690’ 임상 3상을 중단하면서 'BBT-877'의 가치가 한층 높아질 수 있다는 기대감도 나타냈다. 브릿지바이오테라퓨틱스 이정규 대표는 이날 온라인 IR 설명회를 개최하고 "FDA가 BBT-877의 안전한 임상이 수행될 수 있도록 추가 시험을 권고한 것에 대해 고무적으로 생각한다"라며 "오토택신 저해제 계열 내 최초 약물로 개발하기 위해 연구활동에 매진하겠다"라고 말했다.2021-06-28 11:25:08안경진
브릿지바이오 "베링거 반환 신약, 추가 독성실험 추진"[데일리팜=안경진 기자] 브릿지바이오테라퓨틱스는 특발성 폐섬유증 치료후보물질 'BBT-877' 개발과 관련해 미국식품의약국(FDA)으로부터 서면 회신을 수령했다고 28일 공시했다. 'BBT-877'의 추가 실험을 거쳐 임상2상 설계를 공고히 하라는 FDA의 권고사항을 받아들여 연말까지 추가 실험을 진행하고 관련 데이터를 확보하겠다는 방침이다. 'BBT-877'은 브릿지바이오가 지난 2019년 7월 베링거인겔하임에 기술이전했다가 작년 11월 권리를 돌려받은 특발성 폐섬유증 신약후보물질이다. 다양한 세포종에서 섬유화를 관할하는 오토택신 효소를 저해하는 기전으로 작용한다. 브릿지바이오가 원개발사인 레고켐바이오로부터 'BBT-877'를 도입한 다음 1상임상 단계에서 기술이전 계약을 체결했는데, 임상 1상 당시 병행한 비임상 실험에서 잠재적 독성 우려가 제기되면서 최종적으로 권리 반환이 결정됐다. 브릿지바이오는 FDA와 지난 3월 'BBT-877' 개발 관련 회의를 진행한지 3개월 여만에 서면으로 FDA 입장을 전달받은 상황이다. 브릿지바이오에 따르면 FDA는 'BBT-877' 관련 In vivo GLP 혜성 분석과 전자현미경을 통한 세포사멸 분석 등의 비임상 데이터를 추가로 제출하라고 권고했다. 브릿지바이오는 자체 및 외부 실험을 통해 약물의 직접적 DNA 손상이 아닌, 고농도 약물 처리로 인한 세포사멸 기전이 'BBT-877'의 잠재적 독성 우려를 발생시킨 원인이라고 보고 있다. 이같은 입장을 FDA에 소명하고, 독성반응이 위양성이라는 사실을 재확인하기 위해 추가 실험을 진행하기로 협의한 것이다. 브릿지바이오는 이번에 수령한 FDA 권고에 따라 ▲생체 내 실험을 통한 혜성 분석(in vivo Comet assay) ▲닌테다닙 및 피르페니돈 등 특발성 폐섬유증 환자에서의 기존 표준 치료제와의 약물상호작용(DDI) 시험에 곧바로 착수하겠다고 밝혔다. 올해 연말까지 두 데이터를 추가로 확보하고, FDA와 추가적인 상의 절차를 거쳐 임상 2상의 최종 설계를 구체화한다는 계획이다. 내년 상반기에는 임상 2상의 시험계획신청(CTA)을 완료하고 임상 개시가 가능할 것으로 내다봤다. 특발성 폐섬유증 외에 진행성 표현형을 나타내는 만성 섬유성 간질성폐질환(PF-ILD) 및 다양한 암종을 타깃하는 적응증 확대 연구도 연내 구체화한다는 방침이다. 올해 초 경쟁사인 갈라파고스가 동일 계열의 신약후보물질 ‘GLPG1690’ 임상 3상을 중단하면서 'BBT-877'의 가치가 한층 높아질 수 있다는 기대감도 나타냈다. 브릿지바이오테라퓨틱스 이정규 대표는 이날 온라인 IR 설명회를 개최하고 "FDA가 BBT-877의 안전한 임상이 수행될 수 있도록 추가 시험을 권고한 것에 대해 고무적으로 생각한다"라며 "오토택신 저해제 계열 내 최초 약물로 개발하기 위해 연구활동에 매진하겠다"라고 말했다.2021-06-28 11:25:08안경진 -
 일동, 미 당뇨병학회서 당뇨신약 전임상결과 발표[데일리팜=김진구 기자] 일동제약이 25~29일(현지시간) 개최되는 2021 미국 당뇨병학회(ADA)에서 당뇨신약 후보물질 'IDG16177'에 대한 전임상 결과를 포스터로 발표했다고 28일 밝혔다. & 8203; IDG16177은 GPR40 Agonist 계열의 신약 후보물질이다. 췌장 베타세포의 GPR40을 활성화해 인슐린 분비를 유도하고 혈당을 조절하는 기전이다. & 8203; 일동제약은 이번 발표에서 간독성 등의 문제로 개발이 중단된 기존의 유사계열 후보물질 '파시글리팜(fasiglifam)'과의 비교연구 결과가 강조됐다고 설명했다. & 8203; 발표에 따르면 전임상시험에서 IDG16177은 체외실험에서 파시글리팜에 비해 더 우수한 활성을 보였다. 파시글리팜보다 100배 낮은 농도에서도 인슐린 분비를 유도하는 것으로 나타났다. & 8203; 동물실험 결과 약동학적으로 약물 흡수가 우수했으며, 파시글리팜 대비 30배 낮은 용량에서도 우수한 혈당조절 능력을 보였다. & 8203; 약물에 의한 간 독성(DILI)을 현저히 낮춘 것으로도 나타났다. IDG16177은 전임상 독성시험을 통해 임상시험에 적용할 수 있는 충분한 안전역을 확보, 인체 유효농도가 낮을 것으로 예상된다는 설명이다. & 8203; 일동제약은 최근 독일연방 의약품·의료기기관리기관(BfArM)에 IDG16177 임상1상 계획 승인을 요청한 상태다. 임상계획이 승인되면 독일에서 임상1상에 돌입하게 된다. & 8203; 일동제약 관계자는 "IDG16177의 혈당강하 등 유효성은 물론, 독성 등 안전성 측면에서 경쟁력을 확인했다"며 "임상시료 확보 등 임상을 위한 제반 준비가 완료된 만큼, 임상계획이 승인되는 대로 신속하게 임상1상에 돌입할 계획"이라고 말했다.2021-06-28 10:48:12김진구
일동, 미 당뇨병학회서 당뇨신약 전임상결과 발표[데일리팜=김진구 기자] 일동제약이 25~29일(현지시간) 개최되는 2021 미국 당뇨병학회(ADA)에서 당뇨신약 후보물질 'IDG16177'에 대한 전임상 결과를 포스터로 발표했다고 28일 밝혔다. & 8203; IDG16177은 GPR40 Agonist 계열의 신약 후보물질이다. 췌장 베타세포의 GPR40을 활성화해 인슐린 분비를 유도하고 혈당을 조절하는 기전이다. & 8203; 일동제약은 이번 발표에서 간독성 등의 문제로 개발이 중단된 기존의 유사계열 후보물질 '파시글리팜(fasiglifam)'과의 비교연구 결과가 강조됐다고 설명했다. & 8203; 발표에 따르면 전임상시험에서 IDG16177은 체외실험에서 파시글리팜에 비해 더 우수한 활성을 보였다. 파시글리팜보다 100배 낮은 농도에서도 인슐린 분비를 유도하는 것으로 나타났다. & 8203; 동물실험 결과 약동학적으로 약물 흡수가 우수했으며, 파시글리팜 대비 30배 낮은 용량에서도 우수한 혈당조절 능력을 보였다. & 8203; 약물에 의한 간 독성(DILI)을 현저히 낮춘 것으로도 나타났다. IDG16177은 전임상 독성시험을 통해 임상시험에 적용할 수 있는 충분한 안전역을 확보, 인체 유효농도가 낮을 것으로 예상된다는 설명이다. & 8203; 일동제약은 최근 독일연방 의약품·의료기기관리기관(BfArM)에 IDG16177 임상1상 계획 승인을 요청한 상태다. 임상계획이 승인되면 독일에서 임상1상에 돌입하게 된다. & 8203; 일동제약 관계자는 "IDG16177의 혈당강하 등 유효성은 물론, 독성 등 안전성 측면에서 경쟁력을 확인했다"며 "임상시료 확보 등 임상을 위한 제반 준비가 완료된 만큼, 임상계획이 승인되는 대로 신속하게 임상1상에 돌입할 계획"이라고 말했다.2021-06-28 10:48:12김진구 -
 동아ST, 미 당뇨병학회에서 신약 임상1b상 결과 발표[데일리팜=김진구 기자] 동아에스티는 25~28일(현지시간) 개최되는 미국 당뇨병학회(ADA 2021)에서 제2형 당뇨병치료제로 개발 중인 'DA-1241'의 미국 임상1b상 결과를 발표했다고 28일 밝혔다. DA-1241은 GPR119(G protein-coupled receptor 119) 작용제 기전의 퍼스트 인 클래스 신약으로 개발 중이다. GPR119는 췌장의 베타세포에 존재하는 수용체로, 활성화되면 포도당·지질 대사 산물의 양에 따라 인슐린 분비를 증가시킨다. DA-1241은 이 수용체를 활성화해 저혈당 위험 없이 식후 혈당을 개선한다. 미국 임상1b상은 정상인과 제2형 당뇨병 환자를 대상으로 반복투여·용량증량 시험으로 진행됐다. 당뇨병 환자들은 메트포르민을 복용하면서 위약과 시타글립틴, 또는 DA-1241 25·50·100mg을 1일 1회 8주간 복용했다. 대조시험·이중눈가림·무작위배정으로 진행된 임상시험에서 DA-1241의 우수한 임상적 유의성이 확인됐다. 식후혈당 감소 효과는 혈당증가분의 곡선하면적(incremental AUE0-4h) 측정으로 평가했다. 복용 전 대비 DA-1241 100mg(-13.8%)이 시타글립틴 100mg(-9.0%)과 유사한 혈당개선을 효과를 나타냈다. 위약(+10.5%) 대비 매우 우수한 혈당개선 효과가 나타났다. 공복혈당과 연속혈당측정을 통한 혈당변동성 지표에서는 시타글립틴과 유사한 수준을 보였다. 특히 복용 시 GLP-1의 분비량이 증가하는 것으로 나타나, 체내에서 DA-1241의 GPR119 수용체 활성화를 확인했다. 반면, 시타글립틴은 복용 후 시간이 경과하면서 GLP-1 분비가 감소하는 것으로 나타났다. 임상적으로 의미 있는 부작용은 관찰되지 않았으며, DA-1241 100mg(-2.2%, -1.57kg)이 위약(-0.3%)과 시타글립틴(-0.3%) 대비 상대적으로 높은 체중 감소효과를 나타냈다. 동아에스티 관계자는 "많은 제약사가 GPR119 작용제 기전 치료제 개발을 진행했으나 임상적 유효성 입증에 실패했다. 반면, 동아에스티는 비임상에서 유효성이 개선된 후보물질을 도출하고 임상에서도 특징을 확인했다"며 "연내에 미국에서 임상2상 IND를 신청할 계획"이라고 말했다. 미국 당뇨병학회는 세계에서 가장 권위 있는 당뇨병 관련 국제학회다. 작년에 이어 올해도 온라인으로 개최됐다. DA-1241의 자세한 미국 임상1b상 결과는 ADA2021 등록자에 한해 학회 웹사이트 내 e-포스터 세션에서 포스터·초록으로 확인할 수 있다.2021-06-28 10:27:32김진구
동아ST, 미 당뇨병학회에서 신약 임상1b상 결과 발표[데일리팜=김진구 기자] 동아에스티는 25~28일(현지시간) 개최되는 미국 당뇨병학회(ADA 2021)에서 제2형 당뇨병치료제로 개발 중인 'DA-1241'의 미국 임상1b상 결과를 발표했다고 28일 밝혔다. DA-1241은 GPR119(G protein-coupled receptor 119) 작용제 기전의 퍼스트 인 클래스 신약으로 개발 중이다. GPR119는 췌장의 베타세포에 존재하는 수용체로, 활성화되면 포도당·지질 대사 산물의 양에 따라 인슐린 분비를 증가시킨다. DA-1241은 이 수용체를 활성화해 저혈당 위험 없이 식후 혈당을 개선한다. 미국 임상1b상은 정상인과 제2형 당뇨병 환자를 대상으로 반복투여·용량증량 시험으로 진행됐다. 당뇨병 환자들은 메트포르민을 복용하면서 위약과 시타글립틴, 또는 DA-1241 25·50·100mg을 1일 1회 8주간 복용했다. 대조시험·이중눈가림·무작위배정으로 진행된 임상시험에서 DA-1241의 우수한 임상적 유의성이 확인됐다. 식후혈당 감소 효과는 혈당증가분의 곡선하면적(incremental AUE0-4h) 측정으로 평가했다. 복용 전 대비 DA-1241 100mg(-13.8%)이 시타글립틴 100mg(-9.0%)과 유사한 혈당개선을 효과를 나타냈다. 위약(+10.5%) 대비 매우 우수한 혈당개선 효과가 나타났다. 공복혈당과 연속혈당측정을 통한 혈당변동성 지표에서는 시타글립틴과 유사한 수준을 보였다. 특히 복용 시 GLP-1의 분비량이 증가하는 것으로 나타나, 체내에서 DA-1241의 GPR119 수용체 활성화를 확인했다. 반면, 시타글립틴은 복용 후 시간이 경과하면서 GLP-1 분비가 감소하는 것으로 나타났다. 임상적으로 의미 있는 부작용은 관찰되지 않았으며, DA-1241 100mg(-2.2%, -1.57kg)이 위약(-0.3%)과 시타글립틴(-0.3%) 대비 상대적으로 높은 체중 감소효과를 나타냈다. 동아에스티 관계자는 "많은 제약사가 GPR119 작용제 기전 치료제 개발을 진행했으나 임상적 유효성 입증에 실패했다. 반면, 동아에스티는 비임상에서 유효성이 개선된 후보물질을 도출하고 임상에서도 특징을 확인했다"며 "연내에 미국에서 임상2상 IND를 신청할 계획"이라고 말했다. 미국 당뇨병학회는 세계에서 가장 권위 있는 당뇨병 관련 국제학회다. 작년에 이어 올해도 온라인으로 개최됐다. DA-1241의 자세한 미국 임상1b상 결과는 ADA2021 등록자에 한해 학회 웹사이트 내 e-포스터 세션에서 포스터·초록으로 확인할 수 있다.2021-06-28 10:27:32김진구 -
 릴리, 변이에 '속수무책'…항체 치료제 美 공급 중단[데일리팜=정새임 기자] 일라이 릴리의 코로나19 항체 칵테일 요법이 변이 바이러스에 충분한 효과를 보이지 못해 공급이 중단됐다. 미국 보건부(HHS)와 식품의약국(FDA)은 지난 25일(현지시간) 자로 릴리의 코로나19 항체 치료제 '밤라니비맙'과 '에테세비맙'의 공급 중단 결정을 내렸다. 인비트로 실험에서 두 치료제를 조합한 칵테일 요법이 브라질(감마 변종)과 남아공(베타 변종)발 코로나19 바이러스 변이에 활성을 나타내지 못했기 때문이다. 미국 질병통제예방센터(CDC)에 따르면 브라질과 남아프리카 변종은 미국 내 전체 감염자의 11% 이상을 차지하며, 그 수가 증가하고 있다. 릴리는 코로나19 대유행 중 가장 먼저 항체 치료제를 선보였지만 변이 바이러스가 유행하면서 초기 효과를 유지하지 못했다. 밤라니비맙 단독요법은 지난 4월 FDA 긴급사용승인이 취소되기도 했다. 미 보건부는 리제네론의 코로나19 항체 칵테일 요법 'REGEN-COV'와 GSK-Vir의 항체 치료제 '소트로비맙'이 변이 바이러스에 효과적이라며 릴리 항체 치료제 대신 두 요법을 사용할 것을 권장했다. 일라이 릴리는 "밤라니비맙과 에테세비맙을 함께 투여하면 감마 혹은 베타 변종에 중화 효과를 유지하지 않는다"고 시인하며 "변이가 진화하고 그 전파 패턴과 유병률이 변화함에 따라 전 세계 정부 및 규제 기관과 협력하여 적절한 환자에게 항체를 제공할 수 있도록 노력할 것"이라고 밝혔다.2021-06-28 10:26:27정새임
릴리, 변이에 '속수무책'…항체 치료제 美 공급 중단[데일리팜=정새임 기자] 일라이 릴리의 코로나19 항체 칵테일 요법이 변이 바이러스에 충분한 효과를 보이지 못해 공급이 중단됐다. 미국 보건부(HHS)와 식품의약국(FDA)은 지난 25일(현지시간) 자로 릴리의 코로나19 항체 치료제 '밤라니비맙'과 '에테세비맙'의 공급 중단 결정을 내렸다. 인비트로 실험에서 두 치료제를 조합한 칵테일 요법이 브라질(감마 변종)과 남아공(베타 변종)발 코로나19 바이러스 변이에 활성을 나타내지 못했기 때문이다. 미국 질병통제예방센터(CDC)에 따르면 브라질과 남아프리카 변종은 미국 내 전체 감염자의 11% 이상을 차지하며, 그 수가 증가하고 있다. 릴리는 코로나19 대유행 중 가장 먼저 항체 치료제를 선보였지만 변이 바이러스가 유행하면서 초기 효과를 유지하지 못했다. 밤라니비맙 단독요법은 지난 4월 FDA 긴급사용승인이 취소되기도 했다. 미 보건부는 리제네론의 코로나19 항체 칵테일 요법 'REGEN-COV'와 GSK-Vir의 항체 치료제 '소트로비맙'이 변이 바이러스에 효과적이라며 릴리 항체 치료제 대신 두 요법을 사용할 것을 권장했다. 일라이 릴리는 "밤라니비맙과 에테세비맙을 함께 투여하면 감마 혹은 베타 변종에 중화 효과를 유지하지 않는다"고 시인하며 "변이가 진화하고 그 전파 패턴과 유병률이 변화함에 따라 전 세계 정부 및 규제 기관과 협력하여 적절한 환자에게 항체를 제공할 수 있도록 노력할 것"이라고 밝혔다.2021-06-28 10:26:27정새임
-
 정규직 2026년 신입 약사 채용
정규직 2026년 신입 약사 채용 -
 5/29(금) 하루 근무하실 약사님 연락부탁드립니다~
5/29(금) 하루 근무하실 약사님 연락부탁드립니다~ -
 한국의약품안전관리원 개방형 직위 (의약품안전조사본부장) 채용 재공고
한국의약품안전관리원 개방형 직위 (의약품안전조사본부장) 채용 재공고 -
 [부산보훈병원] '26년 정규직 약사 공개채용
[부산보훈병원] '26년 정규직 약사 공개채용 -
 CTD담당/국내 RA 경력 채용
CTD담당/국내 RA 경력 채용 -
 [강동성심병원] 약사 채용공고(ASP전담 / 정규직 약사 / 주말약사)
[강동성심병원] 약사 채용공고(ASP전담 / 정규직 약사 / 주말약사) -
 경남 함안공장 제조관리약사 책임자 채용
경남 함안공장 제조관리약사 책임자 채용 -
 약제부 주말(토,일 각각)전담 약사 모집
약제부 주말(토,일 각각)전담 약사 모집 -
 Specialist, Hematology and Oncology Marketing
Specialist, Hematology and Oncology Marketing -
 경력 및 신입 약사 모집
경력 및 신입 약사 모집 -
 해외사업본부 PIS팀 약사 채용
해외사업본부 PIS팀 약사 채용 -
 계명대학교 동산의료원 2026학년도 임시직(야간약사) 계약직원 상시 공개채용 공고
계명대학교 동산의료원 2026학년도 임시직(야간약사) 계약직원 상시 공개채용 공고 -
 약제팀 (협약직) 야간약사 모집
약제팀 (협약직) 야간약사 모집 -
 BD/마케팅PM(소화기/근골격/항생제) 경력 채용
BD/마케팅PM(소화기/근골격/항생제) 경력 채용 -
 제조관리 약사 채용 신입/경력
제조관리 약사 채용 신입/경력 -
 [서울 30분] 약제센터 야간 약사모집
[서울 30분] 약제센터 야간 약사모집 -
 동원아이팜 관리약사 채용
동원아이팜 관리약사 채용 -
 대외협력실 PR(Public Relations) 경력사원(계약직) 채용
대외협력실 PR(Public Relations) 경력사원(계약직) 채용 -
 제제연구/개선/건기식 개발 경력 채용
제제연구/개선/건기식 개발 경력 채용 -
Senior Manager, Government Affairs & External Liaison
-
 관리약사/주사제 제조/영업 담당자 모집
관리약사/주사제 제조/영업 담당자 모집 -
 일산차병원 약제팀 주간약사 채용
일산차병원 약제팀 주간약사 채용 -
 [대자인병원] 약제팀 약사 / ASP팀 감염전문약사 모집
[대자인병원] 약제팀 약사 / ASP팀 감염전문약사 모집 -
 RPT 약리/의약화학 연구원 모집
RPT 약리/의약화학 연구원 모집 -
 KGSP_헬스케어 품질 및 규정 관리 담당자(과~차장급)_정규직 채용
KGSP_헬스케어 품질 및 규정 관리 담당자(과~차장급)_정규직 채용 -
 [중앙보훈병원] 약제실 약사 채용
[중앙보훈병원] 약제실 약사 채용 -
 약제팀 토요일 시간제 약사 모집
약제팀 토요일 시간제 약사 모집 -
 원주 공장 관리약사 채용 경력무관(신입우대)
원주 공장 관리약사 채용 경력무관(신입우대) -
 토/일/공휴일 당직약사님 모집
토/일/공휴일 당직약사님 모집 -
 로뎀요양병원 약사님 모십니다.
로뎀요양병원 약사님 모십니다. -
 2026년 제28회 직원모집(약사_팀장) 공고
2026년 제28회 직원모집(약사_팀장) 공고 -
 ★ 약제과 오후 파트 정규직 약사 초빙 ★
★ 약제과 오후 파트 정규직 약사 초빙 ★ -
 계약직 신입직원 [한방조제팀 한약사] 공개채용 모집
계약직 신입직원 [한방조제팀 한약사] 공개채용 모집 -
 한국에자이 QA Associate 채용
한국에자이 QA Associate 채용 -
 '정규직 약사' '야간약사' '주말약사' 채용
'정규직 약사' '야간약사' '주말약사' 채용



오늘의 TOP 10
- 1제네릭 약가 산정률 45%…혁신형·준혁신형·수급안정, 약가우대
- 2유상준 약학정보원장 직위해제…임명 1년 2개월 만
- 3[단독] 상비약 자판기 규제특례 재추진…"차기 회의서 결판"
- 4휴텍스제약, 제네릭 약가재평가 소송 최종 승소…"약가인하 부당"
- 5기등재 인하 1·2차 갈림길...'지각생동·복합제' 구제 관건
- 6린버크 물질특허 회피 심판 청구…우판권 물거품 가능성
- 7명인제약, 영업익 첫 1천억 돌파 보인다…CNS 1위 질주
- 8여름 비염, 오래가는 코막힘…'점막 염증 관리' 중요한 이유
- 9미래바이오 생산 7개 제품 품질 부적합 우려 전량 회수
- 10복지부 1차관에 현수엽 대변인…"현장경험과 전문성 겸비"