



총 23건
-
 움트, 임상시험 수탁사업 총괄 선덕성 대표 영입[데일리팜=이탁순 기자] 움트는 임상시험 수탁사업(CRO) 강화를 위해 선덕성 신임 대표를 영입했다고 6일 밝혔다. 이번 인사는 움트가 CRO 시장에서 본격적인 성장 궤도에 진입하겠다는 의지를 보여주는 전략적 결정으로 평가된다. 신임 선덕성 대표는 CRO 사업을 총괄하는 각자 대표로, 기존 신남철 대표는 회사 경영 전반과 신약개발 사업을 지휘한다. 선덕성 신임 대표는 CRO 업계에서 검증된 경영 전문가다. 코스닥 상장사인 디티앤씨알오(Dt&CRO)와 현대ADM에서 총괄 사장을 역임하며 두 회사의 성장을 이끌었고, 바이오코아에서는 BD 업무를 담당하며 기업공개(IPO) 성공에 기여했다. 비임상부터 임상시험까지 신약개발 전 과정에 대한 실무 경험을 갖춘 그의 합류는 움트의 CRO 사업 역량을 한 단계 끌어올릴 것으로 기대된다. 움트는 선 대표의 영입과 함께 국내외 제약사 및 바이오벤처와의 협력 네트워크를 대폭 확대할 계획이다. 특히 Phase 1부터 Phase 4까지 모든 단계의 임상시험을 지원하는 전주기 서비스 체계를 강화하고, 연구자주도 임상시험 분야에서 쌓아온 식약처 인허가 노하우를 적극 활용해 시장 점유율을 높여나갈 방침이다. 현재 움트의 CRO 사업은 프로토콜 개발부터 IRB 및 식약처 IND 절차 지원, 데이터 관리, 통계 분석, 임상시험 보고서(CSR) 작성까지 임상시험의 전 과정을 커버하고 있다. 의약품은 물론 의료기기, 체외진단의료기기 임상시험까지 수행하며 폭넓은 서비스 포트폴리오를 구축했다. 특히 국내 의료계와의 광범위한 연구자 네트워크는 움트의 핵심 경쟁력으로 꼽힌다. 움트는 CRO 사업 외에도 헬스케어 커뮤니케이션 분야에서 독보적인 위치를 차지하고 있다. 'eyefit', 'Liver Update', 'Join OS', ‘Womb story’, 'HeartBit', ‘Bonejour’, ‘URO World’, ‘Mind Up’등 13종 이상의 진료과별 의료정보 매거진을 발행하며 의료계와 긴밀한 관계를 유지하고 있다. 이러한 의료계 네트워크는 임상시험 수주와 연구자 확보에 큰 자산이 되고 있다는 설명이다. 2023년 움트는 사명의 의미를 'Unique Medical-service Traders'로 재정의하며 독창적인 토탈 의료 솔루션을 공급하고 연결하는 회사로 거듭났다. 임상시험 수탁, 신약 개발, 메디컬 마케팅, 의료정보 제공 등 헬스케어 산업 전반을 아우르는 종합 의료 솔루션 기업으로 포지셔닝한 것이다. 신남철 대표는 "선덕성 신임 대표의 합류로 지속 가능한 CRO로 한 단계 성장하는 기틀을 마련하게 되었다"며 기대감을 표했다. 회사 관계자는 "이번 인사는 CRO 사업의 도약을 위한 중요한 전환점이 될 것"이라며 "임상시험 품질과 서비스 역량을 지속적으로 강화해 고객사와의 신뢰를 구축해 나가겠다"고 밝혔다. 업계에서는 움트의 이번 인사를 긍정적으로 평가하고 있다. 검증된 경영진 영입, 전주기 임상시험 서비스 체계, 광범위한 의료계 네트워크, 신약 개발 파이프라인 등이 맞물려 시너지를 낼 것으로 전망된다. 특히 국내 CRO 시장이 연평균 10% 이상 성장하고 있는 가운데, 움트의 경쟁력 강화는 시장 점유율 확대로 이어질 것으로 보인다. 2004년 설립한 움트는 서울 구로구 에이스테크노타워에 본사를 두고 있다. 회사 관계자는 "선덕성 신임 대표 체제 하에서 국내외 제약사 및 바이오벤처와의 협력을 확대하고 임상시험 분야 경쟁력을 더욱 강화해 나갈 계획"이라고 강조했다.2026-01-06 10:52:50이탁순 기자
움트, 임상시험 수탁사업 총괄 선덕성 대표 영입[데일리팜=이탁순 기자] 움트는 임상시험 수탁사업(CRO) 강화를 위해 선덕성 신임 대표를 영입했다고 6일 밝혔다. 이번 인사는 움트가 CRO 시장에서 본격적인 성장 궤도에 진입하겠다는 의지를 보여주는 전략적 결정으로 평가된다. 신임 선덕성 대표는 CRO 사업을 총괄하는 각자 대표로, 기존 신남철 대표는 회사 경영 전반과 신약개발 사업을 지휘한다. 선덕성 신임 대표는 CRO 업계에서 검증된 경영 전문가다. 코스닥 상장사인 디티앤씨알오(Dt&CRO)와 현대ADM에서 총괄 사장을 역임하며 두 회사의 성장을 이끌었고, 바이오코아에서는 BD 업무를 담당하며 기업공개(IPO) 성공에 기여했다. 비임상부터 임상시험까지 신약개발 전 과정에 대한 실무 경험을 갖춘 그의 합류는 움트의 CRO 사업 역량을 한 단계 끌어올릴 것으로 기대된다. 움트는 선 대표의 영입과 함께 국내외 제약사 및 바이오벤처와의 협력 네트워크를 대폭 확대할 계획이다. 특히 Phase 1부터 Phase 4까지 모든 단계의 임상시험을 지원하는 전주기 서비스 체계를 강화하고, 연구자주도 임상시험 분야에서 쌓아온 식약처 인허가 노하우를 적극 활용해 시장 점유율을 높여나갈 방침이다. 현재 움트의 CRO 사업은 프로토콜 개발부터 IRB 및 식약처 IND 절차 지원, 데이터 관리, 통계 분석, 임상시험 보고서(CSR) 작성까지 임상시험의 전 과정을 커버하고 있다. 의약품은 물론 의료기기, 체외진단의료기기 임상시험까지 수행하며 폭넓은 서비스 포트폴리오를 구축했다. 특히 국내 의료계와의 광범위한 연구자 네트워크는 움트의 핵심 경쟁력으로 꼽힌다. 움트는 CRO 사업 외에도 헬스케어 커뮤니케이션 분야에서 독보적인 위치를 차지하고 있다. 'eyefit', 'Liver Update', 'Join OS', ‘Womb story’, 'HeartBit', ‘Bonejour’, ‘URO World’, ‘Mind Up’등 13종 이상의 진료과별 의료정보 매거진을 발행하며 의료계와 긴밀한 관계를 유지하고 있다. 이러한 의료계 네트워크는 임상시험 수주와 연구자 확보에 큰 자산이 되고 있다는 설명이다. 2023년 움트는 사명의 의미를 'Unique Medical-service Traders'로 재정의하며 독창적인 토탈 의료 솔루션을 공급하고 연결하는 회사로 거듭났다. 임상시험 수탁, 신약 개발, 메디컬 마케팅, 의료정보 제공 등 헬스케어 산업 전반을 아우르는 종합 의료 솔루션 기업으로 포지셔닝한 것이다. 신남철 대표는 "선덕성 신임 대표의 합류로 지속 가능한 CRO로 한 단계 성장하는 기틀을 마련하게 되었다"며 기대감을 표했다. 회사 관계자는 "이번 인사는 CRO 사업의 도약을 위한 중요한 전환점이 될 것"이라며 "임상시험 품질과 서비스 역량을 지속적으로 강화해 고객사와의 신뢰를 구축해 나가겠다"고 밝혔다. 업계에서는 움트의 이번 인사를 긍정적으로 평가하고 있다. 검증된 경영진 영입, 전주기 임상시험 서비스 체계, 광범위한 의료계 네트워크, 신약 개발 파이프라인 등이 맞물려 시너지를 낼 것으로 전망된다. 특히 국내 CRO 시장이 연평균 10% 이상 성장하고 있는 가운데, 움트의 경쟁력 강화는 시장 점유율 확대로 이어질 것으로 보인다. 2004년 설립한 움트는 서울 구로구 에이스테크노타워에 본사를 두고 있다. 회사 관계자는 "선덕성 신임 대표 체제 하에서 국내외 제약사 및 바이오벤처와의 협력을 확대하고 임상시험 분야 경쟁력을 더욱 강화해 나갈 계획"이라고 강조했다.2026-01-06 10:52:50이탁순 기자 -
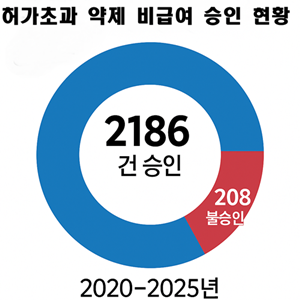 허가초과 약제 비급여 승인, 6년간 불승인 10배 넘어[데일리팜=정흥준 기자] 지난 6년간 허가초과 약제(오프라벨) 비급여 사용 승인 건이 불승인 건수의 10배가 넘는 것으로 나타났다. 허가초과 사용 신청이 208건 불승인되는 동안 위험성 대비 유익성이 상회한다는 등의 이유로 2186건이 승인됐다. 건강보험심사평가원은 27일 일반약제를 포함한 허가초과 약제 비급여 승인 건수를 처음 공개했다. 오프라벨 약제의 비급여 사용 신청은 IRB(생명윤리위원회)가 설치된 병원 또는 학회에서 심의 후 가능하다. 일반 약제는 심평원이 식약처에 의뢰해 안전성·유효성을 검토하고, 항암제는 매달 열리는 암질환심의위원회에서 승인하고 있다. 항암제와 달리 일반약제는 그동안 불승인 내역만 공개해왔다. 이에 따라 요양기관들이 오프라벨 비급여 사용 신청을 할 때 어려움을 겪는다는 문제가 지적돼 왔다. 또 형평성에 어긋나기 때문에 일반약제도 승인 내역을 공개해야 한다는 요청이 있었다. 심평원은 지난 9월 ‘허가초과 약제 비급여 사용 승인업무 운영규정 일부개정안’을 예고하고 의견 조회를 진행한 바 있다. 이번에 공개된 승인사례는 2020년부터 현재까지 2186건으로, 2020년 472건, 2021년 572건, 2022년 266건, 2023년 485건, 2024년 276건, 올해 115건이다. 또 불승인 사례는 2013년부터 2025년까지 13년 동안 377건이 공개됐다. 승인사례가 공개된 2020년부터는 208건이 불승인됐다. 연도별 불승인 추이를 살펴보면 2020년 33건, 2021년 86건, 2022년 38건, 2023년 44건, 2024년 6건, 올해 1건이다. 작년과 올해 불승인 사례가 크게 줄어든 것을 확인할 수 있다. 공개된 불승인 사례 자료에서는 올해 맙테라주(리툭시맙) 불승인 1건이 추가됐다. 대체약제보다 비용 효과적이거나 부작용이 적고, 치료효과가 높을 것으로 기대된다는 이유로 신청했으나 제출된 근거자료로 확인되지 않는다는 사유다.2025-10-27 11:26:40정흥준
허가초과 약제 비급여 승인, 6년간 불승인 10배 넘어[데일리팜=정흥준 기자] 지난 6년간 허가초과 약제(오프라벨) 비급여 사용 승인 건이 불승인 건수의 10배가 넘는 것으로 나타났다. 허가초과 사용 신청이 208건 불승인되는 동안 위험성 대비 유익성이 상회한다는 등의 이유로 2186건이 승인됐다. 건강보험심사평가원은 27일 일반약제를 포함한 허가초과 약제 비급여 승인 건수를 처음 공개했다. 오프라벨 약제의 비급여 사용 신청은 IRB(생명윤리위원회)가 설치된 병원 또는 학회에서 심의 후 가능하다. 일반 약제는 심평원이 식약처에 의뢰해 안전성·유효성을 검토하고, 항암제는 매달 열리는 암질환심의위원회에서 승인하고 있다. 항암제와 달리 일반약제는 그동안 불승인 내역만 공개해왔다. 이에 따라 요양기관들이 오프라벨 비급여 사용 신청을 할 때 어려움을 겪는다는 문제가 지적돼 왔다. 또 형평성에 어긋나기 때문에 일반약제도 승인 내역을 공개해야 한다는 요청이 있었다. 심평원은 지난 9월 ‘허가초과 약제 비급여 사용 승인업무 운영규정 일부개정안’을 예고하고 의견 조회를 진행한 바 있다. 이번에 공개된 승인사례는 2020년부터 현재까지 2186건으로, 2020년 472건, 2021년 572건, 2022년 266건, 2023년 485건, 2024년 276건, 올해 115건이다. 또 불승인 사례는 2013년부터 2025년까지 13년 동안 377건이 공개됐다. 승인사례가 공개된 2020년부터는 208건이 불승인됐다. 연도별 불승인 추이를 살펴보면 2020년 33건, 2021년 86건, 2022년 38건, 2023년 44건, 2024년 6건, 올해 1건이다. 작년과 올해 불승인 사례가 크게 줄어든 것을 확인할 수 있다. 공개된 불승인 사례 자료에서는 올해 맙테라주(리툭시맙) 불승인 1건이 추가됐다. 대체약제보다 비용 효과적이거나 부작용이 적고, 치료효과가 높을 것으로 기대된다는 이유로 신청했으나 제출된 근거자료로 확인되지 않는다는 사유다.2025-10-27 11:26:40정흥준 -
 퓨처켐 "루도타다이펩, 조건부 허가 연내 신청"[베를린 2025 ESMO=황병우 기자=황병우 기자] 유럽임상종양학회(ESMO 2025)에서 공개된 퓨쳐켐의 차세대 방사성리간드치료제(RLT) ‘루도타다이펩(FC705)’ 결과가 주목받고 있다. 전이성 거세저항성 전립선암(mCRPC) 환자를 대상으로 한 다회 투여 임상에서 주요 장기의 누적 방사선량이 국제 안전 기준을 하회하며, 반복 투여 시에도 내약성을 유지하는 것으로 확인됐다. 데일리팜은 현지 베를린에서 박찬수 퓨쳐켐 개발본부 이사를 만나 이번 결과의 의미와 향후 개발 전략을 들어봤다. "알부민 결합 구조로 체내 순환 연장…안전성 우려 해소" 박 이사는 "알부민 결합형 구조로 체내 반감기를 늘리면서 종양 축적은 높이고, 정상 장기 피폭은 증가하지 않는다는 점을 이번 데이터로 입증했다"고 밝혔다. 이번 연구는 국내 임상 2상으로, mCRPC 환자 20명을 대상으로 총 68회의 치료 주기를 분석했다 . 첫 투여 시 침샘(1.22±0.53 Gy/GBq)과 신장(0.674±0.33 Gy/GBq)에서 가장 높은 흡수선량을 보였고, 골수(0.053±0.011 Gy/GBq)는 가장 낮았다. 반복 투여 후 침샘 선량은 유의하게 감소(p2025-10-21 12:00:28황병우
퓨처켐 "루도타다이펩, 조건부 허가 연내 신청"[베를린 2025 ESMO=황병우 기자=황병우 기자] 유럽임상종양학회(ESMO 2025)에서 공개된 퓨쳐켐의 차세대 방사성리간드치료제(RLT) ‘루도타다이펩(FC705)’ 결과가 주목받고 있다. 전이성 거세저항성 전립선암(mCRPC) 환자를 대상으로 한 다회 투여 임상에서 주요 장기의 누적 방사선량이 국제 안전 기준을 하회하며, 반복 투여 시에도 내약성을 유지하는 것으로 확인됐다. 데일리팜은 현지 베를린에서 박찬수 퓨쳐켐 개발본부 이사를 만나 이번 결과의 의미와 향후 개발 전략을 들어봤다. "알부민 결합 구조로 체내 순환 연장…안전성 우려 해소" 박 이사는 "알부민 결합형 구조로 체내 반감기를 늘리면서 종양 축적은 높이고, 정상 장기 피폭은 증가하지 않는다는 점을 이번 데이터로 입증했다"고 밝혔다. 이번 연구는 국내 임상 2상으로, mCRPC 환자 20명을 대상으로 총 68회의 치료 주기를 분석했다 . 첫 투여 시 침샘(1.22±0.53 Gy/GBq)과 신장(0.674±0.33 Gy/GBq)에서 가장 높은 흡수선량을 보였고, 골수(0.053±0.011 Gy/GBq)는 가장 낮았다. 반복 투여 후 침샘 선량은 유의하게 감소(p2025-10-21 12:00:28황병우 -
 심평원, 약제 허가 초과 제도 개선 본격화…자문단과 논의[데일리팜=이탁순 기자] 건강보험심사평가원(원장 강중구, 이하 심평원)이 약제 허가 초과 제도 개선에 본격적으로 나섰다. 심평원은 11일 미래전략위원회 회의를 개최하고, 약제 및 치료재료 허가범위 초과사용과 관련한 제도 개선 방안을 논의했다고 밝혔다. 지난 2023년 12월 출범한 미래전략위원회는 의약단체, 언론계, 시민단체, 보건의료계 등 해당분야의 경험과 학식이 풍부한 전문가로 구성돼 심사평가제도 발전 및 미래비전 전략 수립 등에 관한 주요의제에 대해 논의하는 심사평가원 자문기구이다. 이번 회의에서는 약제 및 치료재료 허가범위 초과사용 평가제도 관련 ▲승인절차 간소화 ▲안전관리 강화 ▲재평가 및 사후관리 개선 등에 대해 이은주 급여관리실장이 보고했다. 이어 위원장인 차의과대학교 전병율 교수를 비롯해 박은철, 서인석, 이상운, 이우용, 서동철, 홍석철, 신성식, 강정화, 이동욱 위원이 다양한 의견을 나누며 활발한 토론을 이어갔다. 특히 위원들은 ▲과도하게 경직된 IRB 심의 절차의 효율성 제고 ▲승인·불승인 결과 공개로 투명성 확보를 통한 이해관계자 신뢰 향상 ▲안전성·효과성에 대한 모니터링 체계 강화 등에 대한 심도 있는 논의를 진행했다는 설명이다. 강중구 심평원장은 "허가범위 초과사용 평가제도를 개선하기 위해 연구용역을 실시했고, 지난 8월 29일 국제 심포지엄을 비롯해 여러 채널을 통해 의견을 수렴하고 검토하고 있다"며, "앞으로도 환자의 안전을 위해 제기된 현안을 차근차근 해결해 나가고, 보다 안전하고 수준 높은 의료환경을 조성해 나가겠다"고 밝혔다.2025-09-12 16:41:07이탁순
심평원, 약제 허가 초과 제도 개선 본격화…자문단과 논의[데일리팜=이탁순 기자] 건강보험심사평가원(원장 강중구, 이하 심평원)이 약제 허가 초과 제도 개선에 본격적으로 나섰다. 심평원은 11일 미래전략위원회 회의를 개최하고, 약제 및 치료재료 허가범위 초과사용과 관련한 제도 개선 방안을 논의했다고 밝혔다. 지난 2023년 12월 출범한 미래전략위원회는 의약단체, 언론계, 시민단체, 보건의료계 등 해당분야의 경험과 학식이 풍부한 전문가로 구성돼 심사평가제도 발전 및 미래비전 전략 수립 등에 관한 주요의제에 대해 논의하는 심사평가원 자문기구이다. 이번 회의에서는 약제 및 치료재료 허가범위 초과사용 평가제도 관련 ▲승인절차 간소화 ▲안전관리 강화 ▲재평가 및 사후관리 개선 등에 대해 이은주 급여관리실장이 보고했다. 이어 위원장인 차의과대학교 전병율 교수를 비롯해 박은철, 서인석, 이상운, 이우용, 서동철, 홍석철, 신성식, 강정화, 이동욱 위원이 다양한 의견을 나누며 활발한 토론을 이어갔다. 특히 위원들은 ▲과도하게 경직된 IRB 심의 절차의 효율성 제고 ▲승인·불승인 결과 공개로 투명성 확보를 통한 이해관계자 신뢰 향상 ▲안전성·효과성에 대한 모니터링 체계 강화 등에 대한 심도 있는 논의를 진행했다는 설명이다. 강중구 심평원장은 "허가범위 초과사용 평가제도를 개선하기 위해 연구용역을 실시했고, 지난 8월 29일 국제 심포지엄을 비롯해 여러 채널을 통해 의견을 수렴하고 검토하고 있다"며, "앞으로도 환자의 안전을 위해 제기된 현안을 차근차근 해결해 나가고, 보다 안전하고 수준 높은 의료환경을 조성해 나가겠다"고 밝혔다.2025-09-12 16:41:07이탁순 -
 심평원, 허가초과 약제 승인여부 내역 모두 공개[데일리팜=이탁순 기자] 앞으로 허가초과 약제(오프라벨) 승인 여부 내역이 모두 홈페이지에 공개된다. 현재 일반약제의 경우 불승인 약제만 공개되고 있는데, 앞으로는 불승인 내역 뿐만 아니라 승인 내역도 공개해 신청 요양기관의 행정적 부담을 경감시킨다는 방침이다. 건강보험심사평가원은 10일 허가 또는 신고범위 초과 약제 비급여 사용 승인업부 운영규정 일부개정규정안을 사전 예고하고하고, 16일까지 의견 조회에 나섰다. 오프라벨 약제의 비급여 사용 신청은 IRB(생명윤리위원회)가 설치된 병원 및 학회에서 심의 후 가능하다. 이 가운데 일반 약제는 심평원이 식약처에 의뢰해 안전성·유효성을 검토하고, 항암제는 매달 열리는 암질환심의위원회에서 승인한다. 문제는 오프라벨 일반약제의 경우 불승인 내역만 홈페이지에 공개해 요양기관들이 정보 미습득으로 신청서 작성시 어려움이 있었다는 것이다. 특히, 오프라벨 항암제의 경우에는 승인내역과 불승인 내역이 모두 공개되고 있는 데 반해 오프라벨 일반약제는 불승인 내역만 공개되면서 형평성 지적도 받았다. 심평원 측은 "이번에 허가초과 약제의 승인 및 불승인 내역을 모두 홈페이징 공개함에 따라 요양기관이 사용 신청서 작성 시 행정적 부담이 경감되길 기대한다"고 밝혔다. 업계에서는 현재 허가초과 승인 기준을 완화해야 한다는 주장이 나오고 있다. 특히 IRB 심의 조건 폐지와 임상 자료 조건을 완화해야 한다는 주장이다. 업계 한 관계자는 "코로나19 같은 감염병 약제의 경우 질환 특성상 임상시험이 어렵지만, 오프라벨 비급여로 사용하려면 일반약제 같은 임상 문헌을 요구한다"며 "이는 긴급히 사용해야 하는 감염병 치료제의 특성을 감안하지 않는 비합리적인 조치"라고 지적했다. 심평원도 작년 외부 연구용역으로 '약제 및 치료재료 허가범위 초과 사용 제도 개선방안'을 마련하고, 지난달 29일에는 관련해 국제 심포지엄을 여는 등 제도 개선 의지를 보이고 있다. 이번 규정 개정은 제도 개선의 첫 단추로, 앞으로 더 많은 개선 방안들이 논의될 것으로 전망된다.2025-09-11 09:46:24이탁순
심평원, 허가초과 약제 승인여부 내역 모두 공개[데일리팜=이탁순 기자] 앞으로 허가초과 약제(오프라벨) 승인 여부 내역이 모두 홈페이지에 공개된다. 현재 일반약제의 경우 불승인 약제만 공개되고 있는데, 앞으로는 불승인 내역 뿐만 아니라 승인 내역도 공개해 신청 요양기관의 행정적 부담을 경감시킨다는 방침이다. 건강보험심사평가원은 10일 허가 또는 신고범위 초과 약제 비급여 사용 승인업부 운영규정 일부개정규정안을 사전 예고하고하고, 16일까지 의견 조회에 나섰다. 오프라벨 약제의 비급여 사용 신청은 IRB(생명윤리위원회)가 설치된 병원 및 학회에서 심의 후 가능하다. 이 가운데 일반 약제는 심평원이 식약처에 의뢰해 안전성·유효성을 검토하고, 항암제는 매달 열리는 암질환심의위원회에서 승인한다. 문제는 오프라벨 일반약제의 경우 불승인 내역만 홈페이지에 공개해 요양기관들이 정보 미습득으로 신청서 작성시 어려움이 있었다는 것이다. 특히, 오프라벨 항암제의 경우에는 승인내역과 불승인 내역이 모두 공개되고 있는 데 반해 오프라벨 일반약제는 불승인 내역만 공개되면서 형평성 지적도 받았다. 심평원 측은 "이번에 허가초과 약제의 승인 및 불승인 내역을 모두 홈페이징 공개함에 따라 요양기관이 사용 신청서 작성 시 행정적 부담이 경감되길 기대한다"고 밝혔다. 업계에서는 현재 허가초과 승인 기준을 완화해야 한다는 주장이 나오고 있다. 특히 IRB 심의 조건 폐지와 임상 자료 조건을 완화해야 한다는 주장이다. 업계 한 관계자는 "코로나19 같은 감염병 약제의 경우 질환 특성상 임상시험이 어렵지만, 오프라벨 비급여로 사용하려면 일반약제 같은 임상 문헌을 요구한다"며 "이는 긴급히 사용해야 하는 감염병 치료제의 특성을 감안하지 않는 비합리적인 조치"라고 지적했다. 심평원도 작년 외부 연구용역으로 '약제 및 치료재료 허가범위 초과 사용 제도 개선방안'을 마련하고, 지난달 29일에는 관련해 국제 심포지엄을 여는 등 제도 개선 의지를 보이고 있다. 이번 규정 개정은 제도 개선의 첫 단추로, 앞으로 더 많은 개선 방안들이 논의될 것으로 전망된다.2025-09-11 09:46:24이탁순 -
 "감염병 약제 허가초과 기준 완화해야…임상 어려워"[데일리팜=이탁순 기자] 의약품의 허가사항을 초과해 사용하는 데 대해 보다 유연성이 필요하다는 지적이 나온다. 사용 조건과 규제가 엄격한데다 절차도 복잡해 환자들이 치료받을 수 있는 기회를 놓칠 수 있다는 것이다. 특히 감염병 치료제의 경우, 요건을 더 완화해 감염병 유행 방지 차원에서 신속한 대응이 필요하다는 의견이 나온다. 지난 29일 건강보험심사평가원은 서울 중구 웨스틴 조선 호텔에서 '약제와 치료재의 허가범위 초과사용 승인제도 현황과 개선방향'을 주제로 국제 심포지엄을 열었다. 우리나라의 경우 허가 초과 약제(오프라벨)를 사용하려면 병원(또는 학회) 내 설치된 IRB(생명윤리위원회) 심의를 통과해 심평원에 신청해야 한다. 신청된 일반 약제일 경우 심평원은 식약처에 의뢰해 안전성·유효성을 검토하고, 항암제일 경우 매달 열리는 암질환심의위원회에서 사용여부를 결정하게 된다. 문제는 일부 대형병원에만 IRB가 설치돼 있어 신청까지 어렵고, 신청한다고 해도 엄격한 기준에 의해 불승인되는 사례가 많다는 것이다. 이날 심포지엄에서는 지난 5월 결과가 발표된 '약제 및 치료재료 허가범위 초과 사용 제도 개선방안 연구(심평원 외부 용역 연구)'에서 책임자로 이름을 올린 서동철 심평원 객원 연구위원이 발제를 맡았다. 그는 "한국을 제외한 해외 5개 주요국은 허가범위 초과 사용을 위해 IRB 승인을 요구하지 않는다"면서 "불필요한 IRB 요건은 생락하고, 환자 보호를 위한 표준동의서 도입 등 보완책을 마련해야 한다"고 주장했다. 아울러 심평원 산하 오프라벨 전담위원회를 설치해 1주일 내 결과를 통보하거나, 사후 보고 제도도 도입해야 한다고 제안했다. 허가 초과 항암제도 최소한의 안전성·유효성을 확보하는 차원에서 기준을 완화해야 한다는 목소리가 나왔다. 이재련 아산병원 종양내과 교수는 '효과적일 수 있는 약제를 시도해 볼 수 있는 자유와 권리'라는 주제 발표에서 "급성 진행암 환자의 경우 초록 발표만 나온 약제 사용도 중요한 근거가 될 수 있으나, 암질심에서는 인정되지 않는다"며 "오히려 암질심이 환자 생존권을 제약하는 과도한 장벽이 됐다"고 지적했다. 이에 다학제위원회 심의를 거친 기관은 암질심 심의를 생략하고, 학회 전문가 의견을 의무적으로 반영해야 한다고 강조했다. 제약업계에서는 감염병 허가 초과 약제의 기준을 완화해야 한다고 주장한다. 현재 허가초과 비급여 승인을 위해서는 임상문헌근거 기준이 RCT 수준을 요구하고 있다. 하지만 법정감염병의 경우 특정 시기나 지역에서만 유행하거나 높은 치명률 및 집당 발생 위험으로 임상시험을 수행하기 어렵다. 이에 관찰연구에서 효과를 확인한 약물일지라도 RCT 수준의 임상시험을 수행하기는 현실적으로 쉽지 않다. 이에 제약업계는 코로나19와 같은 법정감염병 약제의 경우 신속한 대응 차원에서 임상문헌근거 기준을 RCT 문헌이 아닌 코호트 문헌 등으로 완화해 개선할 필요가 있다고 주장하고 있다. 이같은 제안은 서동철 위원이 연구해 5월 발표한 '약제 및 치료재료 허가범위 초과 사용 제도 개선방안 연구'에도 언급돼 있다. 연구에서는 "법정 감염병 예방 및 치료약제의 경우, 여러 관찰연구에서 감염병 예방 및 치료에 효과를 확인한 약물일지라도 감염병 특성상 특정 시기나 지역에서만 유행하거나, 높은 치명률 및 집단 발생의 위험이 존재하는 등 임상시험을 수행하기 어려운 조건이므로, 해당 약제에 한해 신청 약제 사용 적절성 평가를 위한 임상 근거 범위 및 기준을 확대할 필요가 있다"고 제안했다. 제약바이오협회도 법정 감영병에 사용되는 약제의 허가범위 초과 비급여 사용 신청 시 임상 근거 기준을 완화해 달라며 정부에 건의서를 제출한 상황이다. 협회 측은 심평원 전담 위원회 설치도 주문했다. 제약업계 관계자는 "각 국가의 상황에 맞춰 적절한 약제 사용으로 환자의 생명을 지키기 위해서는 안전성이 충분히 입증된 감염병 약제의 경우 신청 약제 사용의 적절성 평가를 위한 임상 근거 범위 및 기준을 완화할 필요가 있다"고 강조했다.2025-08-31 16:18:49이탁순
"감염병 약제 허가초과 기준 완화해야…임상 어려워"[데일리팜=이탁순 기자] 의약품의 허가사항을 초과해 사용하는 데 대해 보다 유연성이 필요하다는 지적이 나온다. 사용 조건과 규제가 엄격한데다 절차도 복잡해 환자들이 치료받을 수 있는 기회를 놓칠 수 있다는 것이다. 특히 감염병 치료제의 경우, 요건을 더 완화해 감염병 유행 방지 차원에서 신속한 대응이 필요하다는 의견이 나온다. 지난 29일 건강보험심사평가원은 서울 중구 웨스틴 조선 호텔에서 '약제와 치료재의 허가범위 초과사용 승인제도 현황과 개선방향'을 주제로 국제 심포지엄을 열었다. 우리나라의 경우 허가 초과 약제(오프라벨)를 사용하려면 병원(또는 학회) 내 설치된 IRB(생명윤리위원회) 심의를 통과해 심평원에 신청해야 한다. 신청된 일반 약제일 경우 심평원은 식약처에 의뢰해 안전성·유효성을 검토하고, 항암제일 경우 매달 열리는 암질환심의위원회에서 사용여부를 결정하게 된다. 문제는 일부 대형병원에만 IRB가 설치돼 있어 신청까지 어렵고, 신청한다고 해도 엄격한 기준에 의해 불승인되는 사례가 많다는 것이다. 이날 심포지엄에서는 지난 5월 결과가 발표된 '약제 및 치료재료 허가범위 초과 사용 제도 개선방안 연구(심평원 외부 용역 연구)'에서 책임자로 이름을 올린 서동철 심평원 객원 연구위원이 발제를 맡았다. 그는 "한국을 제외한 해외 5개 주요국은 허가범위 초과 사용을 위해 IRB 승인을 요구하지 않는다"면서 "불필요한 IRB 요건은 생락하고, 환자 보호를 위한 표준동의서 도입 등 보완책을 마련해야 한다"고 주장했다. 아울러 심평원 산하 오프라벨 전담위원회를 설치해 1주일 내 결과를 통보하거나, 사후 보고 제도도 도입해야 한다고 제안했다. 허가 초과 항암제도 최소한의 안전성·유효성을 확보하는 차원에서 기준을 완화해야 한다는 목소리가 나왔다. 이재련 아산병원 종양내과 교수는 '효과적일 수 있는 약제를 시도해 볼 수 있는 자유와 권리'라는 주제 발표에서 "급성 진행암 환자의 경우 초록 발표만 나온 약제 사용도 중요한 근거가 될 수 있으나, 암질심에서는 인정되지 않는다"며 "오히려 암질심이 환자 생존권을 제약하는 과도한 장벽이 됐다"고 지적했다. 이에 다학제위원회 심의를 거친 기관은 암질심 심의를 생략하고, 학회 전문가 의견을 의무적으로 반영해야 한다고 강조했다. 제약업계에서는 감염병 허가 초과 약제의 기준을 완화해야 한다고 주장한다. 현재 허가초과 비급여 승인을 위해서는 임상문헌근거 기준이 RCT 수준을 요구하고 있다. 하지만 법정감염병의 경우 특정 시기나 지역에서만 유행하거나 높은 치명률 및 집당 발생 위험으로 임상시험을 수행하기 어렵다. 이에 관찰연구에서 효과를 확인한 약물일지라도 RCT 수준의 임상시험을 수행하기는 현실적으로 쉽지 않다. 이에 제약업계는 코로나19와 같은 법정감염병 약제의 경우 신속한 대응 차원에서 임상문헌근거 기준을 RCT 문헌이 아닌 코호트 문헌 등으로 완화해 개선할 필요가 있다고 주장하고 있다. 이같은 제안은 서동철 위원이 연구해 5월 발표한 '약제 및 치료재료 허가범위 초과 사용 제도 개선방안 연구'에도 언급돼 있다. 연구에서는 "법정 감염병 예방 및 치료약제의 경우, 여러 관찰연구에서 감염병 예방 및 치료에 효과를 확인한 약물일지라도 감염병 특성상 특정 시기나 지역에서만 유행하거나, 높은 치명률 및 집단 발생의 위험이 존재하는 등 임상시험을 수행하기 어려운 조건이므로, 해당 약제에 한해 신청 약제 사용 적절성 평가를 위한 임상 근거 범위 및 기준을 확대할 필요가 있다"고 제안했다. 제약바이오협회도 법정 감영병에 사용되는 약제의 허가범위 초과 비급여 사용 신청 시 임상 근거 기준을 완화해 달라며 정부에 건의서를 제출한 상황이다. 협회 측은 심평원 전담 위원회 설치도 주문했다. 제약업계 관계자는 "각 국가의 상황에 맞춰 적절한 약제 사용으로 환자의 생명을 지키기 위해서는 안전성이 충분히 입증된 감염병 약제의 경우 신청 약제 사용의 적절성 평가를 위한 임상 근거 범위 및 기준을 완화할 필요가 있다"고 강조했다.2025-08-31 16:18:49이탁순 -
![[칼럼] 의료기기 '선진입-후평가' 실효성 제고 개선 방향](https://pds.dailypharm.com/news_image/202508/325726_1.jpg) [칼럼] 의료기기 '선진입-후평가' 실효성 제고 개선 방향최근 국내 의료기기 제도는 기술 혁신을 뒷받침하는 방향으로 진일보하고 있다. 특히 '선진입-후평가' 제도는 안전성이 입증된 의료기기의 신속한 시장 진입을 허용하고, 이후 실제 사용 데이터를 기반으로 유효성을 평가하는 체계를 구축했다. 이는 전 세계적인 기술 경쟁에서 국내 의료기기산업이 경쟁력을 확보하는 데 중요한 제도적 발판이 되고 있다. 로봇과 인공지능(AI) 등 융합 기술을 기반으로 한 혁신 의료기기들이 시장에서 안착한다면, 이는 단순한 산업 발전을 넘어 국가의 고부가가치 성장 동력으로 자리매김할 수 있다. 그러나 이 제도가 효과적으로 작동해 실질적인 임상적 성과와 산업적 성장을 이끌기 위해서는 운영상의 여러 문제를 짚고 넘어갈 필요가 있다. 로엔서지컬의 선진입 사례에서 본 문제점 필자는 국내 수술로봇 제조기업 로엔서지컬의 공동창업자이자 이사로, 수술로봇 '자메닉스'의 개발과 실사용을 주도하고 있다. 이 과정에서 직접 경험한 선진입-후평가 제도의 한계와 현실적인 애로사항은 제도 개선의 필요성을 절감하게 만들었다. 로엔서지컬은 비뇨기과 수술로봇 자메닉스를 통해 고위험 신장 질환 환자들에게 새로운 치료 옵션을 제공하고 있다. 해당 기기는 ‘혁신의료기술’로 지정되면서 선진입 제도에 들어섰지만, 실제 환자에게 적용되기까지는 1년이 넘는 시간이 걸렸다. 이는 임상연구계획에 대한 한국보건의료연구원 심의 승인, 실시기관 사용신고, 장비 도입, 임시수가코드 생성 등 다수의 절차를 진행해야만 하기 때문이다. 이로 인해 고시된 3년의 선진입 기간 중 약 1/3은 실제 환자 적용이 불가능했고, 남는 기간동안 필수적인 임상 데이터를 충분이 축적하는 데 어려움이 발생했다. 이에 더해, 비침습적 의료기기와 달리 침습 기술이 적용된 의료기기는 혁신의료기술 선진입 제도에서 근거창출연구 목적 이외 임상 진료가 사실상 제한돼 있으며, 이는 임상 사례를 충분히 축적하기 어려운 구조적 제약이 된다. 혁신의료기술 실시에 관한 지침에 따르면, 근거창출연구 중이라도 연구계획서상 목표 등록 환자수 모집 종료 시 임상 진료로 전환이 가능하다. 그러나 실제 현장에서 환자 등록은 수술 완료 이후에야 가능하기 때문에, 실질적으로 ‘모집 종료’ 시점은 ‘연구 종료’ 시점과 동일하게 적용된다. 게다가 근거창출연구는 대부분 3차 의료기관에서 수행되는데, 이들 기관은 기저질환이나 중증 질환을 가진 환자가 많아, 상황에 따라 연구 기준에 적합한 대상자 모집이 쉽지 않다. 이는 진료 전환까지의 시간 지연요소가 되고, 결과적으로 실사용 데이터 확보에 어려움을 초래한다. 침습적 혁신의료기술의 유효성과 임상적 가치를 평가·입증하기 위해서는 엄격히 통제된 임상연구 외에도, 실제 사용 환경에서 다양한 환자군을 대상으로 한 근거 수집이 병행돼야 한다. 따라서 일정 조건을 충족한 침습적 혁신의료기술에 대해서는, IRB 승인 기관을 중심으로 진료와 연구를 병행할 수 있도록 유연한 제도 운영이 필요하다. 이를 통해 환자에게 보다 확대된 치료 선택권을 제공함과 동시에, 제도의 궁극적 목표인 기술의 안전성과 유효성 검증 확보에도 실질적인 기여를 할 수 있을 것이다. 제도 개선을 위한 네 가지 방향 로엔서지컬의 사례 외에도, 큐렉소, 엘엔로보틱스 등 국산 침습 의료기기를 개발하고 있는 기업들은 유사한 문제를 답습하고 있다. 이런 문제 해결을 위해 현장에서 체감하는 선진입 제도의 운영상 문제에 대해 다음 네가지 측면에서 제도적 개선방안을 제안하고자 한다. 첫째는 심의 절차의 신속성 확보다. 현재 근거창출연구 심의는 월 1회로 제한돼 있고, 기업과 근거·평가위원회 간 직접 소통 채널의 부재로, 연구 계획의 경미한 변경에도 많은 시간이 소요된다. 기업이 실제 연구 일정을 원활히 운영하기 위한 기업-위원회 소통 채널 구축과 연구에 미치는 영향이 경미한 변경 사항의 경우 간소화된 심의 절차 마련이 필요하다. 둘째는 사용기간의 유연성 확보다. 제도적으로 혁신의료기술로 지정된 이후 연구계획 심의나 장비 도입, 기관 승인 등으로 실제 환자 적용까지 지연된 경우, 이에 대한 기업의 소명 등 근거 축적에 필요한 사용기간 연장을 요청할 수 있는 체계가 필요하다. 셋째는 침습적 혁신의료기술의 임상진료 병행 허용이다. 물론 이는 무조건적인 허용이 아닌 납득할 만한 일정 조건을 충족하는 경우에 한한 허용이다. 예를 들어 모집 목표 환자의 일부가 충족되고 사용현황 보고를 통해 안전성을 확보한 경우 근거창출연구와 임상진료를 일정부분 병행할 수 있도록 한다면 보다 유연한 제도 운용을 기대할 수 있을 것이다. 넷째는 시행 절차의 통합 가이드 마련이다. 한국보건의료연구원, 건강보험심사평가원 등 각 기관마다 행정 절차가 진행되면서, 제도를 이용하는 기업과 병원은 혼란을 겪고 있다. 절차에 대한 이해도를 높이고 접근성을 향상시키기 위해 단계별 매뉴얼을 제공하고 통합 창구를 구축하는 것이 시급하다. 마무리하며 의료기기는 환자의 생명과 직결되는 만큼 안전성과 유효성은 의료기기의 기본 전제다. 이런 기본 전제 위에서 제도의 유연성과 현실성이 함께 뒷받침될 때, 혁신 기술은 비로소 실질적인 진료 현장에 진입하고 이는 의료서비스 질 제고로 이어질 수 있다. 지금도 정부와 유관기관들이 혁신 의료기기산업을 육성하기 위해 끊임없이 정책적 노력을 하고 있으며, 이에 대한 감사를 전한다. 오늘의 제도 혁신은 곧 내일의 환자 치료 기회로 이어진다고 믿는다. 국내 의료기기산업이 단순히 글로벌 트렌드를 따라가는 수준에 머무르지 않고, 세계 시장을 선도하는 수준으로 도약할 수 있도록 제도의 실효성을 높이는 개선이 지속되기를 기대한다. 또한 제도 개선의 한 축으로서, 현장의 목소리가 국내 의료기기산업의 글로벌 성장을 위한 제도 설계와 운영에 반영되기를 희망한다.2025-08-18 08:57:33데일리팜
[칼럼] 의료기기 '선진입-후평가' 실효성 제고 개선 방향최근 국내 의료기기 제도는 기술 혁신을 뒷받침하는 방향으로 진일보하고 있다. 특히 '선진입-후평가' 제도는 안전성이 입증된 의료기기의 신속한 시장 진입을 허용하고, 이후 실제 사용 데이터를 기반으로 유효성을 평가하는 체계를 구축했다. 이는 전 세계적인 기술 경쟁에서 국내 의료기기산업이 경쟁력을 확보하는 데 중요한 제도적 발판이 되고 있다. 로봇과 인공지능(AI) 등 융합 기술을 기반으로 한 혁신 의료기기들이 시장에서 안착한다면, 이는 단순한 산업 발전을 넘어 국가의 고부가가치 성장 동력으로 자리매김할 수 있다. 그러나 이 제도가 효과적으로 작동해 실질적인 임상적 성과와 산업적 성장을 이끌기 위해서는 운영상의 여러 문제를 짚고 넘어갈 필요가 있다. 로엔서지컬의 선진입 사례에서 본 문제점 필자는 국내 수술로봇 제조기업 로엔서지컬의 공동창업자이자 이사로, 수술로봇 '자메닉스'의 개발과 실사용을 주도하고 있다. 이 과정에서 직접 경험한 선진입-후평가 제도의 한계와 현실적인 애로사항은 제도 개선의 필요성을 절감하게 만들었다. 로엔서지컬은 비뇨기과 수술로봇 자메닉스를 통해 고위험 신장 질환 환자들에게 새로운 치료 옵션을 제공하고 있다. 해당 기기는 ‘혁신의료기술’로 지정되면서 선진입 제도에 들어섰지만, 실제 환자에게 적용되기까지는 1년이 넘는 시간이 걸렸다. 이는 임상연구계획에 대한 한국보건의료연구원 심의 승인, 실시기관 사용신고, 장비 도입, 임시수가코드 생성 등 다수의 절차를 진행해야만 하기 때문이다. 이로 인해 고시된 3년의 선진입 기간 중 약 1/3은 실제 환자 적용이 불가능했고, 남는 기간동안 필수적인 임상 데이터를 충분이 축적하는 데 어려움이 발생했다. 이에 더해, 비침습적 의료기기와 달리 침습 기술이 적용된 의료기기는 혁신의료기술 선진입 제도에서 근거창출연구 목적 이외 임상 진료가 사실상 제한돼 있으며, 이는 임상 사례를 충분히 축적하기 어려운 구조적 제약이 된다. 혁신의료기술 실시에 관한 지침에 따르면, 근거창출연구 중이라도 연구계획서상 목표 등록 환자수 모집 종료 시 임상 진료로 전환이 가능하다. 그러나 실제 현장에서 환자 등록은 수술 완료 이후에야 가능하기 때문에, 실질적으로 ‘모집 종료’ 시점은 ‘연구 종료’ 시점과 동일하게 적용된다. 게다가 근거창출연구는 대부분 3차 의료기관에서 수행되는데, 이들 기관은 기저질환이나 중증 질환을 가진 환자가 많아, 상황에 따라 연구 기준에 적합한 대상자 모집이 쉽지 않다. 이는 진료 전환까지의 시간 지연요소가 되고, 결과적으로 실사용 데이터 확보에 어려움을 초래한다. 침습적 혁신의료기술의 유효성과 임상적 가치를 평가·입증하기 위해서는 엄격히 통제된 임상연구 외에도, 실제 사용 환경에서 다양한 환자군을 대상으로 한 근거 수집이 병행돼야 한다. 따라서 일정 조건을 충족한 침습적 혁신의료기술에 대해서는, IRB 승인 기관을 중심으로 진료와 연구를 병행할 수 있도록 유연한 제도 운영이 필요하다. 이를 통해 환자에게 보다 확대된 치료 선택권을 제공함과 동시에, 제도의 궁극적 목표인 기술의 안전성과 유효성 검증 확보에도 실질적인 기여를 할 수 있을 것이다. 제도 개선을 위한 네 가지 방향 로엔서지컬의 사례 외에도, 큐렉소, 엘엔로보틱스 등 국산 침습 의료기기를 개발하고 있는 기업들은 유사한 문제를 답습하고 있다. 이런 문제 해결을 위해 현장에서 체감하는 선진입 제도의 운영상 문제에 대해 다음 네가지 측면에서 제도적 개선방안을 제안하고자 한다. 첫째는 심의 절차의 신속성 확보다. 현재 근거창출연구 심의는 월 1회로 제한돼 있고, 기업과 근거·평가위원회 간 직접 소통 채널의 부재로, 연구 계획의 경미한 변경에도 많은 시간이 소요된다. 기업이 실제 연구 일정을 원활히 운영하기 위한 기업-위원회 소통 채널 구축과 연구에 미치는 영향이 경미한 변경 사항의 경우 간소화된 심의 절차 마련이 필요하다. 둘째는 사용기간의 유연성 확보다. 제도적으로 혁신의료기술로 지정된 이후 연구계획 심의나 장비 도입, 기관 승인 등으로 실제 환자 적용까지 지연된 경우, 이에 대한 기업의 소명 등 근거 축적에 필요한 사용기간 연장을 요청할 수 있는 체계가 필요하다. 셋째는 침습적 혁신의료기술의 임상진료 병행 허용이다. 물론 이는 무조건적인 허용이 아닌 납득할 만한 일정 조건을 충족하는 경우에 한한 허용이다. 예를 들어 모집 목표 환자의 일부가 충족되고 사용현황 보고를 통해 안전성을 확보한 경우 근거창출연구와 임상진료를 일정부분 병행할 수 있도록 한다면 보다 유연한 제도 운용을 기대할 수 있을 것이다. 넷째는 시행 절차의 통합 가이드 마련이다. 한국보건의료연구원, 건강보험심사평가원 등 각 기관마다 행정 절차가 진행되면서, 제도를 이용하는 기업과 병원은 혼란을 겪고 있다. 절차에 대한 이해도를 높이고 접근성을 향상시키기 위해 단계별 매뉴얼을 제공하고 통합 창구를 구축하는 것이 시급하다. 마무리하며 의료기기는 환자의 생명과 직결되는 만큼 안전성과 유효성은 의료기기의 기본 전제다. 이런 기본 전제 위에서 제도의 유연성과 현실성이 함께 뒷받침될 때, 혁신 기술은 비로소 실질적인 진료 현장에 진입하고 이는 의료서비스 질 제고로 이어질 수 있다. 지금도 정부와 유관기관들이 혁신 의료기기산업을 육성하기 위해 끊임없이 정책적 노력을 하고 있으며, 이에 대한 감사를 전한다. 오늘의 제도 혁신은 곧 내일의 환자 치료 기회로 이어진다고 믿는다. 국내 의료기기산업이 단순히 글로벌 트렌드를 따라가는 수준에 머무르지 않고, 세계 시장을 선도하는 수준으로 도약할 수 있도록 제도의 실효성을 높이는 개선이 지속되기를 기대한다. 또한 제도 개선의 한 축으로서, 현장의 목소리가 국내 의료기기산업의 글로벌 성장을 위한 제도 설계와 운영에 반영되기를 희망한다.2025-08-18 08:57:33데일리팜 -
 삼아아토크 임상재평가 연장...2028년까지 결과 제출[데일리팜=이혜경 기자] 삼아제약의 '삼아아토크건조시럽(포르모테롤푸마르산염)' 임상재평가가 24개월 연장된다. 13일 식품의약품안전처가 공개한 중앙약사심의위원회 회의록을 보면 지난 7월 25일 포르모테롤푸마르산염수화물 제제 임상재평가 결과자료 제출기한 연장 사유 및 연장기간의 타당성 여부에 대한 자문이 있었다. 결과적으로는 참석위원 7명 전원 동의로 삼아제약이 제출한 제출기한의 연장 사유와 연장기간(24개월) 산출근거의 적정성이 인정됐다. 삼아제약은 IRB 승인 및 개시 준비 4개월, 대상환자 모집 17개월, 투약 및 추적관찰 1개월, 통계분석 및 보고서 작성 7개월 등 총 29개월의 시간이 소요된다고 밝혔다. 중앙약심 결과로 재평가 자료 제출 기한은 기존 2026년 1월 13일에서 24개월 연장해 2028년 1월 13일까지로 변경된다. 다만 중앙약심 위원들은 기간 연장의 필요성 및 산출기간에는 동의하지만 대상 환자가 소아 급성기관지염 환자라는 점에서 현장에서 임상시험 대상환자 등록이 계획대로 되지 않을 수도 있다는 점을 우려했다. 삼아제약이 신청한 자료제출 24개월 연장이 짧을 수도 있다는 것이다. 한 위원은 "소아 대상 임상시험에서 대상자 선정·제외 기준을 만족하기 어려운 경우가 많다"며 "해당 의약품은 패취제, 흡입제에 비해 용량 조절이 용이하고 소아과에서 반드시 필요한 의약품이므로 임상시험을 완료하여 유의한 결과 도출 필요하다"고 했다. 또 다른 위원도 "연장기간 2년이 임상시험 완료를 위한 최소한의 기간이라고 생각되며, 필요시 업체가 임상시험 실시기관을 추가 확대하는 방안도 고려해야 한다"고 덧붙였다. 한편 식약처는 지난 2020년 12월 23일 포르모테롤푸마르산염 성분 정제·시럽제 등 16품목에 대한 임상재평가를 공고했으며, 1986년 최초 허가 품목인 삼아제약의 삼아아토크정, 삼아아토크건조시럽과 삼아아토크정20마이크로그램 등 3품목을 제외하고 나머지 품목은 모두 허가를 자진 취하했다. 이 과정에서 삼아제약 또한 정제에 대한 생산을 중단하고 건조시럽제에 대한 임상재평가만 진행하기로 결정했다. 삼아제약은 임상재평가 당시 3개 적응증 중 최초 기관지천식, 급만성 기관지염에 대해 임상시험 계획서를 제출했으나, 보완자료 미제출 등으로 인해 행정처분 조치 이후 급성기관지염에 대해서만 임상시험을 진행 중이다. 임상재평가를 진행하면서 2023년 허가사항도 시럽제·정제 급성기관지염의 기도폐쇄성 장애에 의한 호흡곤란 등 여러 증상의 완화로 축소됐다.2025-08-13 17:44:57이혜경
삼아아토크 임상재평가 연장...2028년까지 결과 제출[데일리팜=이혜경 기자] 삼아제약의 '삼아아토크건조시럽(포르모테롤푸마르산염)' 임상재평가가 24개월 연장된다. 13일 식품의약품안전처가 공개한 중앙약사심의위원회 회의록을 보면 지난 7월 25일 포르모테롤푸마르산염수화물 제제 임상재평가 결과자료 제출기한 연장 사유 및 연장기간의 타당성 여부에 대한 자문이 있었다. 결과적으로는 참석위원 7명 전원 동의로 삼아제약이 제출한 제출기한의 연장 사유와 연장기간(24개월) 산출근거의 적정성이 인정됐다. 삼아제약은 IRB 승인 및 개시 준비 4개월, 대상환자 모집 17개월, 투약 및 추적관찰 1개월, 통계분석 및 보고서 작성 7개월 등 총 29개월의 시간이 소요된다고 밝혔다. 중앙약심 결과로 재평가 자료 제출 기한은 기존 2026년 1월 13일에서 24개월 연장해 2028년 1월 13일까지로 변경된다. 다만 중앙약심 위원들은 기간 연장의 필요성 및 산출기간에는 동의하지만 대상 환자가 소아 급성기관지염 환자라는 점에서 현장에서 임상시험 대상환자 등록이 계획대로 되지 않을 수도 있다는 점을 우려했다. 삼아제약이 신청한 자료제출 24개월 연장이 짧을 수도 있다는 것이다. 한 위원은 "소아 대상 임상시험에서 대상자 선정·제외 기준을 만족하기 어려운 경우가 많다"며 "해당 의약품은 패취제, 흡입제에 비해 용량 조절이 용이하고 소아과에서 반드시 필요한 의약품이므로 임상시험을 완료하여 유의한 결과 도출 필요하다"고 했다. 또 다른 위원도 "연장기간 2년이 임상시험 완료를 위한 최소한의 기간이라고 생각되며, 필요시 업체가 임상시험 실시기관을 추가 확대하는 방안도 고려해야 한다"고 덧붙였다. 한편 식약처는 지난 2020년 12월 23일 포르모테롤푸마르산염 성분 정제·시럽제 등 16품목에 대한 임상재평가를 공고했으며, 1986년 최초 허가 품목인 삼아제약의 삼아아토크정, 삼아아토크건조시럽과 삼아아토크정20마이크로그램 등 3품목을 제외하고 나머지 품목은 모두 허가를 자진 취하했다. 이 과정에서 삼아제약 또한 정제에 대한 생산을 중단하고 건조시럽제에 대한 임상재평가만 진행하기로 결정했다. 삼아제약은 임상재평가 당시 3개 적응증 중 최초 기관지천식, 급만성 기관지염에 대해 임상시험 계획서를 제출했으나, 보완자료 미제출 등으로 인해 행정처분 조치 이후 급성기관지염에 대해서만 임상시험을 진행 중이다. 임상재평가를 진행하면서 2023년 허가사항도 시럽제·정제 급성기관지염의 기도폐쇄성 장애에 의한 호흡곤란 등 여러 증상의 완화로 축소됐다.2025-08-13 17:44:57이혜경 -
 지노믹트리 '얼리텍-B' 식약처 허가 최종 자료 제출[데일리팜=이석준 기자] 지노믹트리(대표이사 안성환)는 방광암 체외진단 제품 ‘얼리텍-B’의 국내 사용허가를 위해 식품의약품안전처(이하 ‘식약처’)에 최종 심사자료를 7월 31일자로 제출했다고 밝혔다. 이번 제출 자료는 식약처의 1차 보완 요청에 따라 수행된 추가 중앙 병리의사 재판독 결과와 일부 보충 임상시험 데이터가 반영된 최종 수정 임상보고서다. 지노믹트리는 앞서 국내외 다기관이 참여한 대규모 확증 임상시험을 통해 ‘얼리텍-B’의 임상적 유효성을 확인하고 이를 기반으로 식약처에 사용허가를 신청했다. 해당 확증임상 결과는 참여 의료진 주도로 국제학술지인 ‘자마 온콜로지(JAMA Oncology)’에 게재돼 진단 정확도 및 기술적 완성도를 국제적으로 평가받은 바 있다. 그러나 허가 심사 과정에서 식약처는 일부 기술자료 보완은 물론 병리조직 슬라이드에 대한 추가 중앙 판독, 그리고 보충적 임상시험 수행을 공식 보완사항으로 요구했다. 이에 지노믹트리는 총 8개 임상기관으로부터 처음부터 다시 IRB 승인 및 시료 반출 절차를 수행했으며, 독립 병리 전문가 2인에 의한 중앙 리뷰를 신규로 진행했다. 보완 요청에 따라 추가 임상시험도 설계 및 수행해 이 모든 데이터를 통합한 최종 임상시험 보고서를 식약처에 제출했다. 그 결과, ‘얼리텍-B’의 성능(민감도 및 특이도)은 기존 확증임상시험을 기반으로 ‘자마 온콜로지(JAMA Oncology)’에 보고된 결과와 동등한 수준으로 분석됐다. 이는 임상시험의 신뢰도와 제품의 임상적 견고성을 다시 한번 입증했다는 평가를 받는다. 안성환 지노믹트리 대표는 “이번 중앙 병리 재판독을 위한 추가 임상시험은, 당초 기존 확증임상시험 결과와 유의미한 차이가 없을 것으로 예측되었던 사항들이었으나 식약처의 보완 요청에 따라 성실히 수행됐다”고 밝혔다. 이어 “신개념 의료기기에 대해서는 임상시험 설계 단계부터 보다 실질적이고 합리적인 지원이 필요하다. 향후에는 보다 신속하고 정교한 평가가 가능하도록 모든 이해 당사자간의 상호 학습과 협의 기반의 제도적 정비가 절실하다”고 강조했다. 한편 이번에 제출된 '얼리텍-B' 최종 보완자료는 신속심사 대상에 해당된다. 회사는 식약처의 조속한 심사 절차 착수를 기대하고 있다.2025-07-31 08:55:46이석준
지노믹트리 '얼리텍-B' 식약처 허가 최종 자료 제출[데일리팜=이석준 기자] 지노믹트리(대표이사 안성환)는 방광암 체외진단 제품 ‘얼리텍-B’의 국내 사용허가를 위해 식품의약품안전처(이하 ‘식약처’)에 최종 심사자료를 7월 31일자로 제출했다고 밝혔다. 이번 제출 자료는 식약처의 1차 보완 요청에 따라 수행된 추가 중앙 병리의사 재판독 결과와 일부 보충 임상시험 데이터가 반영된 최종 수정 임상보고서다. 지노믹트리는 앞서 국내외 다기관이 참여한 대규모 확증 임상시험을 통해 ‘얼리텍-B’의 임상적 유효성을 확인하고 이를 기반으로 식약처에 사용허가를 신청했다. 해당 확증임상 결과는 참여 의료진 주도로 국제학술지인 ‘자마 온콜로지(JAMA Oncology)’에 게재돼 진단 정확도 및 기술적 완성도를 국제적으로 평가받은 바 있다. 그러나 허가 심사 과정에서 식약처는 일부 기술자료 보완은 물론 병리조직 슬라이드에 대한 추가 중앙 판독, 그리고 보충적 임상시험 수행을 공식 보완사항으로 요구했다. 이에 지노믹트리는 총 8개 임상기관으로부터 처음부터 다시 IRB 승인 및 시료 반출 절차를 수행했으며, 독립 병리 전문가 2인에 의한 중앙 리뷰를 신규로 진행했다. 보완 요청에 따라 추가 임상시험도 설계 및 수행해 이 모든 데이터를 통합한 최종 임상시험 보고서를 식약처에 제출했다. 그 결과, ‘얼리텍-B’의 성능(민감도 및 특이도)은 기존 확증임상시험을 기반으로 ‘자마 온콜로지(JAMA Oncology)’에 보고된 결과와 동등한 수준으로 분석됐다. 이는 임상시험의 신뢰도와 제품의 임상적 견고성을 다시 한번 입증했다는 평가를 받는다. 안성환 지노믹트리 대표는 “이번 중앙 병리 재판독을 위한 추가 임상시험은, 당초 기존 확증임상시험 결과와 유의미한 차이가 없을 것으로 예측되었던 사항들이었으나 식약처의 보완 요청에 따라 성실히 수행됐다”고 밝혔다. 이어 “신개념 의료기기에 대해서는 임상시험 설계 단계부터 보다 실질적이고 합리적인 지원이 필요하다. 향후에는 보다 신속하고 정교한 평가가 가능하도록 모든 이해 당사자간의 상호 학습과 협의 기반의 제도적 정비가 절실하다”고 강조했다. 한편 이번에 제출된 '얼리텍-B' 최종 보완자료는 신속심사 대상에 해당된다. 회사는 식약처의 조속한 심사 절차 착수를 기대하고 있다.2025-07-31 08:55:46이석준 -
 새 정부 AI 육성 국정기조, 의약품안전관리원도 발맞춘다[데일리팜=이혜경 기자] 이재명 정부의 국정과제 중 하나인 '모두의 인공지능(AI)' 실현이 의약품안전관리체계에도 접목될 전망이다. 한국의약품안전관리원은 새 정부 국정기조에 맞게 수요자인 국민이 체감할 수 있는 실질적인 혁신을 위한 5대 과제를 계획했다. 지난 3월 취임한 손수정 의약품안전관리원장은 15일 식품의약품안전처 출입 전문지 기자단과 만나 "의약품 안전관리체계 선진화, 마약류 오남용 안전관리 강화, 의약품 사용안전망 구축, 혁신기반 미래성장 동력 확보 4대 전략 실행을 위한 5대 과제를 최근 식약처에 보고했다"고 밝혔다. 우선 새 정부의 AI 국정과제 추진에 발맞춰 의약품 안전관리체계를 자동화 기반의 빅데이터·AI 시스템으로 전환해 실사용 정보에 기반한 능동적 약물감시체계를 강화하겠다고 했다. 이를 위해 거대언어모델(LLM)을 활용한 부작용 보고의 국내·외 허가사항 자동 검토 및 인과관계 효율화 분석방법을 개발해 국민에게 신속하게 안전정보를 제공할 수 있도록 2025년부터 3개년 계획으로 정보화 사업과 연계해 단계적으로 추진할 계획이다. 전국 의료기관의 전자의무기록(EMR)을 표준화한 공통데이터모델(CDM)도 현재 30개 기관에서 66개로 대폭 확대해 능동감시 기반을 강화하고, 비정형 자료까지 최대한 연계해 의약품 부작용 모니터링의 정확성을 높일 예정이다. 의약품안전원은 지난 2012년 개원해 13년 동안 국내 의약품 안전관리을 책임지고 있다. 그 결과 의약품 부작용 보고 건수는 153만건으로, 누적 기준 세계 2위 수준이며 30개 병원 EMR을 활용한 CDM을 통해 3896만명, 전 국민 약 75% 수준의 의료정보 기반을 구축했다. 의약품 부작용 피해구제 제도의 경우 현재까지 총 161건, 18억4000만원의 보상금이 지급됐다. 의료용 마약류 안전관리 분야에서는 펜타닐 처방 이력이 있는 160개 병원 처방 소프트웨어 및 2885개 병의원 대상으로 의료쇼핑방지정보망을 연계해 지난해 펜타닐 패치제 처방 14% 감소로 약제비 약 2억8000만원 이상이 절감되는 효과를 도출했다. 손 원장은 "두 번째 과제로 안전한 의약품 사용 환경 마련을 위해 사각지대 없는 의약품 적정사용(DUR) 정보를 개발 제공하고, 챗봇 기반의 국민 친화적 정보 제공 시스템 도입과 임부·노인·만성질환자 등 취약계층을 고려한 안내 자료도 제공을 진행할 것"이라고 했다. 피해자 입장에서 체감할 수 있는 신청 절차 간소화, 디지털 기반 상담 도입, 모바일 안전카드 전환 등 다양한 개선안을 마련해 올해부터 시행에 들어간 것이다. 이와 더불어 의약품 부작용 피해구제제도 개선, 중앙IRB 운영, NIMS 빅데이터를 기반으로 한 실마리 정보 제공 등을 올해 과제로 손꼽았다. 행안부, 법무부, 보건복지부 정보 등과 연계한 지능형 관리시스템 '마약류 오남용 통합감시시스템(K-NASS)'을 2026년까지 구축해 의료용 마약류 오남용·불법 사용 사전 탐지를 통한 실마리 정보를 제공하게 된다. ADHD 치료제, 졸피뎀 등 마약류 투약내역 확인 성분을 순차적으로 확대해 의료쇼핑방지정보망의 적용범위를 넓히고, 행정안전부 국민비서서비스와 연계해 환자가 본인의 마약류 투약내역을 쉽고 안전하게 확인할 수 있는 기반 제공을 추진할 예정이다. 의약품 안전정보 관리시스템을 클라우드 기반의 대국민 서비스, 행정관리 시스템으로 2025년부터 2027년까지 단계적으로 구축·적용할 계획이다. 중앙IRB 공동심사 협약 기관 수를 2025년 90개 이상으로 확대하고, IRB 위원의 전문성 정기 평가, 전문가 풀 확충 및 맞춤형 교육을 통한 임상시험 참여자 권리보호를 위한 전문 상담 역량 강화, 환자 적시 치료기회 제공 등을 위한 임상시험 신속·전문 통합심사 체계 내실화로 안전망 구축 등도 함께 진행하게 된다. 손 원장은 "코로나 팬데믹 이후 전세계적으로 건강에 대한 국민 인식 향상과 부작용 보고 인지도 확산으로 국내·외 시판 후 부작용 보고 자료 증가와 더불어 신속한 분석 평가 제공의 중요성이 부각되고 있다"며 " AI 등 기술을 활용한 의약품안전관리 시스템으로 약물감시 RWD 빅데이터를 자동 분석할 수 있는 시스템 개발과 거대언어모델(LLM) 등 인공지능(AI)을 활용함으로써 의약품 안전정보 관리와 업무 효율화 사업을 계획하고 있다"고 강조했다. 지난 2021년 시범운영으로 시작한 의약품이상사례 보고시스템(KAERS)에 대한 설명도 있었다. KAERS는 올해 3월 의약품안전나라 의약품통합정보시스템에 전면 통합하면서 이상사례 보고서식을 국제표준(ICH E2B(R3))에 따라 개선했다. 그 결과, 국내외 이상사례 보고자료 수집실적이 2020년 147만4000건에서 2024년 152만 8000건으로 증가했다. 제조·수입업체의 의약품 부작용 원시자료 제공서비스의 경우 원시자료 신청건수가 2020년 139건에서 2024년 7622건으로 급증했으나, 자동 제공 시스템 기능 도입으로 평균 제공 소요 시간을 2020년 32일에서 2024년 1일로 대폭 단축했다. 의약품안전원은 전국 28개 의료기관이나 관련 협회·단체를 지역의약품안전센터로 지정해 이상사례 보고 업무를 수행하고 있으며, 지역센터는 의사, 약사 등 의약전문가들로 구성된 상태다. 손 원장은 "지역센터를 통해 국내 누적 이상사례 보고의 약 70%가 이뤄지고 있다"며 "지난해 기준 국내 이상사례 보고는 25만여건으로 전세계 2위에 해당한다"고 밝혔다. 의약품 피해구제제도의 경우 최근 5년간 평균 부담금 징수액은 약 50억원으로, 평균 급여지급액은 약 21억원(42%) 수준이다. 피해구제 신청 건수는 2020년 167건에서 2024년 240건으로 매년 증가추세를 보이고 있으며, 의약품안전원은 인지도 조사 등을 통해 활성화 될 수 있도록 홍보를 강화할 계획이다. 손 원장은 "부작용 피해자는 약물복용 자체로 이미 정신적·신체적 어려움을 겪고 있는데, 제도 접근 장벽까지 높아선 안 된다"며 "AI 기반 상담·처리 체계를 도입해 신청 문턱을 낮추고, 실질적 보상으로 이어지도록 하겠다"고 설명했다.2025-07-16 17:35:25이혜경
새 정부 AI 육성 국정기조, 의약품안전관리원도 발맞춘다[데일리팜=이혜경 기자] 이재명 정부의 국정과제 중 하나인 '모두의 인공지능(AI)' 실현이 의약품안전관리체계에도 접목될 전망이다. 한국의약품안전관리원은 새 정부 국정기조에 맞게 수요자인 국민이 체감할 수 있는 실질적인 혁신을 위한 5대 과제를 계획했다. 지난 3월 취임한 손수정 의약품안전관리원장은 15일 식품의약품안전처 출입 전문지 기자단과 만나 "의약품 안전관리체계 선진화, 마약류 오남용 안전관리 강화, 의약품 사용안전망 구축, 혁신기반 미래성장 동력 확보 4대 전략 실행을 위한 5대 과제를 최근 식약처에 보고했다"고 밝혔다. 우선 새 정부의 AI 국정과제 추진에 발맞춰 의약품 안전관리체계를 자동화 기반의 빅데이터·AI 시스템으로 전환해 실사용 정보에 기반한 능동적 약물감시체계를 강화하겠다고 했다. 이를 위해 거대언어모델(LLM)을 활용한 부작용 보고의 국내·외 허가사항 자동 검토 및 인과관계 효율화 분석방법을 개발해 국민에게 신속하게 안전정보를 제공할 수 있도록 2025년부터 3개년 계획으로 정보화 사업과 연계해 단계적으로 추진할 계획이다. 전국 의료기관의 전자의무기록(EMR)을 표준화한 공통데이터모델(CDM)도 현재 30개 기관에서 66개로 대폭 확대해 능동감시 기반을 강화하고, 비정형 자료까지 최대한 연계해 의약품 부작용 모니터링의 정확성을 높일 예정이다. 의약품안전원은 지난 2012년 개원해 13년 동안 국내 의약품 안전관리을 책임지고 있다. 그 결과 의약품 부작용 보고 건수는 153만건으로, 누적 기준 세계 2위 수준이며 30개 병원 EMR을 활용한 CDM을 통해 3896만명, 전 국민 약 75% 수준의 의료정보 기반을 구축했다. 의약품 부작용 피해구제 제도의 경우 현재까지 총 161건, 18억4000만원의 보상금이 지급됐다. 의료용 마약류 안전관리 분야에서는 펜타닐 처방 이력이 있는 160개 병원 처방 소프트웨어 및 2885개 병의원 대상으로 의료쇼핑방지정보망을 연계해 지난해 펜타닐 패치제 처방 14% 감소로 약제비 약 2억8000만원 이상이 절감되는 효과를 도출했다. 손 원장은 "두 번째 과제로 안전한 의약품 사용 환경 마련을 위해 사각지대 없는 의약품 적정사용(DUR) 정보를 개발 제공하고, 챗봇 기반의 국민 친화적 정보 제공 시스템 도입과 임부·노인·만성질환자 등 취약계층을 고려한 안내 자료도 제공을 진행할 것"이라고 했다. 피해자 입장에서 체감할 수 있는 신청 절차 간소화, 디지털 기반 상담 도입, 모바일 안전카드 전환 등 다양한 개선안을 마련해 올해부터 시행에 들어간 것이다. 이와 더불어 의약품 부작용 피해구제제도 개선, 중앙IRB 운영, NIMS 빅데이터를 기반으로 한 실마리 정보 제공 등을 올해 과제로 손꼽았다. 행안부, 법무부, 보건복지부 정보 등과 연계한 지능형 관리시스템 '마약류 오남용 통합감시시스템(K-NASS)'을 2026년까지 구축해 의료용 마약류 오남용·불법 사용 사전 탐지를 통한 실마리 정보를 제공하게 된다. ADHD 치료제, 졸피뎀 등 마약류 투약내역 확인 성분을 순차적으로 확대해 의료쇼핑방지정보망의 적용범위를 넓히고, 행정안전부 국민비서서비스와 연계해 환자가 본인의 마약류 투약내역을 쉽고 안전하게 확인할 수 있는 기반 제공을 추진할 예정이다. 의약품 안전정보 관리시스템을 클라우드 기반의 대국민 서비스, 행정관리 시스템으로 2025년부터 2027년까지 단계적으로 구축·적용할 계획이다. 중앙IRB 공동심사 협약 기관 수를 2025년 90개 이상으로 확대하고, IRB 위원의 전문성 정기 평가, 전문가 풀 확충 및 맞춤형 교육을 통한 임상시험 참여자 권리보호를 위한 전문 상담 역량 강화, 환자 적시 치료기회 제공 등을 위한 임상시험 신속·전문 통합심사 체계 내실화로 안전망 구축 등도 함께 진행하게 된다. 손 원장은 "코로나 팬데믹 이후 전세계적으로 건강에 대한 국민 인식 향상과 부작용 보고 인지도 확산으로 국내·외 시판 후 부작용 보고 자료 증가와 더불어 신속한 분석 평가 제공의 중요성이 부각되고 있다"며 " AI 등 기술을 활용한 의약품안전관리 시스템으로 약물감시 RWD 빅데이터를 자동 분석할 수 있는 시스템 개발과 거대언어모델(LLM) 등 인공지능(AI)을 활용함으로써 의약품 안전정보 관리와 업무 효율화 사업을 계획하고 있다"고 강조했다. 지난 2021년 시범운영으로 시작한 의약품이상사례 보고시스템(KAERS)에 대한 설명도 있었다. KAERS는 올해 3월 의약품안전나라 의약품통합정보시스템에 전면 통합하면서 이상사례 보고서식을 국제표준(ICH E2B(R3))에 따라 개선했다. 그 결과, 국내외 이상사례 보고자료 수집실적이 2020년 147만4000건에서 2024년 152만 8000건으로 증가했다. 제조·수입업체의 의약품 부작용 원시자료 제공서비스의 경우 원시자료 신청건수가 2020년 139건에서 2024년 7622건으로 급증했으나, 자동 제공 시스템 기능 도입으로 평균 제공 소요 시간을 2020년 32일에서 2024년 1일로 대폭 단축했다. 의약품안전원은 전국 28개 의료기관이나 관련 협회·단체를 지역의약품안전센터로 지정해 이상사례 보고 업무를 수행하고 있으며, 지역센터는 의사, 약사 등 의약전문가들로 구성된 상태다. 손 원장은 "지역센터를 통해 국내 누적 이상사례 보고의 약 70%가 이뤄지고 있다"며 "지난해 기준 국내 이상사례 보고는 25만여건으로 전세계 2위에 해당한다"고 밝혔다. 의약품 피해구제제도의 경우 최근 5년간 평균 부담금 징수액은 약 50억원으로, 평균 급여지급액은 약 21억원(42%) 수준이다. 피해구제 신청 건수는 2020년 167건에서 2024년 240건으로 매년 증가추세를 보이고 있으며, 의약품안전원은 인지도 조사 등을 통해 활성화 될 수 있도록 홍보를 강화할 계획이다. 손 원장은 "부작용 피해자는 약물복용 자체로 이미 정신적·신체적 어려움을 겪고 있는데, 제도 접근 장벽까지 높아선 안 된다"며 "AI 기반 상담·처리 체계를 도입해 신청 문턱을 낮추고, 실질적 보상으로 이어지도록 하겠다"고 설명했다.2025-07-16 17:35:25이혜경 -
 "AI 신약개발 핵심, 데이터 확보...국민신약배당 필요"◆방송: DP초대석 ◆기획: 제약바이오산업2팀 김진구 기자 ◆진행: 제약바이오산업2팀 차지현 기자 ◆촬영·편집: 영상제작팀 ◆출연: 김화종 K-MELLODDY 사업단장 차지현(이하 차): 안녕하세요. 헬스케어 산업 내 다양한 인물을 만나 이야기를 나눠보는 DP 초대석 시간입니다. 오늘은 인공지능 기술과 바이오 산업의 융합을 통해 신약 개발의 새 패러다임을 열고 있는 K-MELLODDY 사업단의 김화종 단장님을 모셨습니다. 단장님, 안녕하세요. 김화종(이하 김): 안녕하세요. K-MELLODDY 사업단 단장을 맡고 있는 김화종입니다. 만나 뵙게 돼서 대단히 반갑습니다. 차: AI 기술이 매우 빠른 속도로 발달하고 있는데요. 최근 미국에서 직접 그 현장을 보고 듣고 오셨다고 들었습니다. 인상 깊었던 장면이 있는지 궁금합니다. 김: 지난 6월, 보스턴에서 열린 바이오USA에 참석했습니다. 130여 개 세션 중 20개 정도가 AI와 관련된 내용이었고요. 크게 세 가지로 나눌 수 있습니다. 첫째는 AI는 빠르게 발전하고 있지만, 바이오 분야에서는 발전이 더딘 편이라는 점입니다. 그 이유는 데이터가 부족하기 때문입니다. 바이오 관련 데이터는 개인 정보가 많고, 병원 데이터나 제약사의 실험 데이터는 외부에 공개되지 않고 있습니다. 둘째는 AI 활용에서 연결의 중요성이 강조됐다는 것입니다. 신약 개발에 AI를 활용할 때, 보통은 단계를 나눠서 활용합니다. 타깃을 찾고 검증하거나, 후보 물질을 찾고 검증하거나, 전임상 데이터를 쓰는 식이죠. 그런데 각 단계를 연결해 사용하는 게 더 효과적입니다. 궁극적으로 임상 1·2·3상을 통과하는 것이 목표이기 때문이죠. 여기에 최근에는 리얼월드 데이터를 AI 모델에 활용해야 한다는 논의도 많습니다. 실험은 매우 제한된 조건에서 이뤄지기 때문에, 실제 사람에게 적용했을 때 잘 맞지 않는 경우가 많습니다. 그래서 FDA에서도 동물실험을 줄이는 방향으로 가고 있고요. 처음부터 다양한 데이터를 함께 학습하는 AI 모델을 만들어야 한다는 얘기가 나왔습니다. 이런 이슈들이 바이오USA에서 핵심적으로 논의된 내용이었습니다. 차: AI 신약 개발 분야에서 K-MELLODDY 사업의 중요성도 점점 커지고 있는 것 같습니다. 이 사업은 어떤 사업이고, 출범한 지 1년 정도 지난 시점에서의 성과는 어떤지 궁금합니다. 김: K-MELLODDY 사업은 제가 한국제약바이오협회 AI신약개발지원센터장을 하던 2021년에 제안한 사업으로, 출범한 지는 4년 정도 됐습니다. 이 사업을 제안한 이유는, 빅데이터의 등장으로 AI 성능이 향상되고 있기 때문입니다. AI는 데이터가 많아야 잘 작동하는데, 신약 개발이나 바이오 분야는 데이터 확보가 굉장히 어렵습니다. 지금 우리가 많이 쓰는 인공지능은 언어모델이나 이미지 인식 분야입니다. 수억 개에서 수십억 개의 데이터를 학습했기 때문이죠. 반면 신약 개발 분야는 수백만 건도 안 되는 경우가 많습니다. 그래서 유럽에서는 10개 제약사가 모여 ‘멜로디(MELLODDY)’라는 프로젝트를 진행했습니다. 경쟁관계에 있지만 데이터를 공동 활용해서 더 좋은 성능의 AI 모델을 만들자는 거죠. 좋은 성능의 모델이 필요한 이유는 실패 확률을 줄이기 위해서입니다. K-MELLODDY는 이 유럽 사례를 참고해 시작됐지만, 구성은 다릅니다. 유럽은 제약사 중심이었다면, 우리는 병원·연구소·대학·창업사까지 모여 더 다양한 데이터를 확보하고자 했습니다. 사업비는 348억 원으로 적지는 않지만, 큰 성과를 내려면 부족한 수준이기도 해서 후속 사업을 고민 중입니다. 지난 1년간은 생태계를 조성하는 데 집중했습니다. 각 기관이 실험 조건과 방식을 공유했고, AI 모델 개발자와 데이터를 보유한 기관이 함께 협력해 모델을 개발하는 작업을 해왔습니다. 데이터가 외부로 나가지 않더라도, 데이터의 구조나 특성을 공유하면서 모델을 만들 수 있는 환경을 구축한 것이 주요 성과입니다. 차: 바이오 업계 사람들과 만나 보면, AI 신약 개발의 필요성은 공감하지만 아직 갈 길이 멀다는 이야기도 많이 합니다. AI 신약 개발에서 가장 중요한 것은 무엇이라고 보시나요? 김: 우리나라 개발자들이나 신약 개발자들의 시각과 글로벌 기업의 시각 차이는 상당히 큽니다. 글로벌은 세계 최고 수준의 인재와 자본이 모여 있기 때문에 첨단 기술이 나올 수밖에 없죠. 그렇다고 우리가 못할 일은 아닙니다. AI 개발에 필요한 기본적인 소프트웨어나 기술은 대부분 공개돼 있습니다. 출발선은 같다고 봐도 돼요. 누가 더 빠르게, 잘 활용하느냐가 관건입니다. 그런데 여기서 늘 문제가 되는 게 데이터입니다. 외국도 지금 데이터를 확보하려고 애쓰고 있고, 우리도 이 분야에 초점을 맞추면 충분히 경쟁력 있는 솔루션을 만들 수 있습니다. 결국 핵심은 ‘데이터 확보’이고, 우리나라도 이 부분은 충분히 할 수 있다고 봅니다. 차: 그렇다면 현재 한국의 AI 신약 개발 수준은 어느 정도라고 판단하시나요? 김: 출발선 자체는 큰 차이가 없습니다. 그런데 실제로는 차이가 큽니다. 예를 들어, AI 모델 정확도가 95%인 것과 96%인 것의 차이는 작아 보이지만, 사람들은 대부분 96%짜리를 선택합니다. 결국 1등 소프트웨어만 사용되는 거죠. 성능만 보면 비슷할 수 있어도, 실제 사용되는 데는 격차가 있습니다. 이걸 좁히기 위해서는 우리만의 무기가 필요합니다. 저는 그것이 ‘잘 정리된 한국의 의료 데이터’라고 생각합니다. 없는 게 아니라, 못 쓰고 있는 거죠. 이 데이터를 잘 활용하면 격차를 줄일 수 있고, 심지어 앞서갈 수도 있다고 생각합니다. 전 국민이 단일 건강보험 체계로 되어 있는 나라는 우리나라와 대만뿐입니다. 이 체계 덕분에 환자가 어떤 치료를 받았는지 쉽게 확인하고 연결할 수 있습니다. 이 데이터를 활용하면, 타깃 발굴, 바이오마커 검증, 약물 반응 추적까지 모두 가능해집니다. 차: 최근 ‘국민신약배당’이라는 새로운 개념을 제안하셨는데요. 어떤 사업인지, 또 어떻게 구상하게 되셨는지 궁금합니다. 김: 제가 ‘국민신약배당’이라는 이름의 사업을 제안했습니다. 이름만 보면 투자사업처럼 들릴 수도 있는데, 일부러 그런 이름을 붙였습니다. 우리나라만이 가진 강점이 바로 ‘잘 정리된 의료 데이터’입니다. 하지만 현재로선 제약이 많습니다. 그래서 이런 생태계를 만들기 위해서는 국민의 이해와 동의가 필요합니다. 지금까지 안 된 이유는 개인정보 보호 때문이라고 하지만, 사실은 개인이 자신의 데이터를 가지고 누군가가 이익을 얻는 것을 쉽게 허용하지 않기 때문이죠. 이 데이터가 공공재로 활용돼야 한다는 것이 제 생각입니다. 그리고 미래에 발생한 이익이 국민에게 배당되는 구조라면, 국민의 저항도 줄어들 겁니다. 좋은 데이터를 기반으로 블록버스터 신약이 만들어지면, 기업이 발전하고, 산업이 성장하고, 다시 국민에게 이익이 돌아가는 선순환이 만들어집니다. 이런 구조를 설명하고자 ‘국민신약배당’이라는 이름을 붙인 겁니다. 차: 국민신약배당 사업이 실제로 이뤄진다면, AI 신약 개발에 큰 도움이 될 것 같습니다. 이 사업을 진행하기 위해 가장 우선적으로 해결해야 할 문제는 무엇이라고 보시나요? 김: 방금 말씀드린 것처럼, 첫 번째는 국민의 동의입니다. 여러 각계각층의 사람들과 이야기해본 결과, 이런 사업이 있으면 좋겠다는 의견이 많았습니다. 산업이 발전하고, 우리나라가 주도하는 블록버스터 신약도 나올 수 있고, 나아가 배당이 이뤄져 건강보험료를 할인받거나 금전적으로 혜택을 얻을 수 있다면, 국민 입장에서도 안 받는 것보다는 낫다는 생각을 하게 될 겁니다. 이런 구조라면 국민 동의를 끌어낼 수 있다고 봅니다. 두 번째는 병원의 협조입니다. 현재는 병원에 가면, 환자들이 연구 목적의 데이터 활용을 대체로 허용하고 있습니다. 하지만 실제 연구는 매우 제한적입니다. IRB(기관생명윤리위원회) 승인을 받아야 하고, 병원 내 데이터만 활용 가능하며, 다른 병원 데이터는 사용할 수 없습니다. 그래서 병원에서도 특정 환자 데이터를 특정 목적에만 제한적으로 쓰던 상황에서, 여러 병원의 데이터를 폭넓게 활용하고, 다양한 AI 모델을 만들어 논문도 쓰고 연구도 할 수 있는 여건이 마련된다면, 오히려 연구 경쟁력을 키울 수 있습니다. 병원도 적극적으로 동참할 수 있는 기반이 생기는 것이죠. 차: 결국 모든 이해관계자들이 마음을 모아야 가능한 사업일 텐데요. 개인이나 병원뿐 아니라, 제약단체나 보건복지부 등 정부 차원의 공감대도 형성되어 있는지 궁금합니다. 김: 네, 사실 이 부분이 가장 중요합니다. 정부나 신약 개발 기업의 적극성이 핵심이죠. 제약기업들은 이런 사업을 환영할 것이라 봅니다. 데이터가 있는 것과 없는 것의 차이는 매우 큽니다. 데이터를 활용하면 신약을 설계하고, 만들고, 검증하는 데 큰 도움이 됩니다. 대신, 제약사들도 신약으로 큰 수익을 낸다면 일정 부분을 공공의 목적으로 환원해야 합니다. 정부 입장에서도 이 사업은 바이오산업을 키우는 중요한 기회입니다. 신약뿐 아니라, 이로 인해 파생되는 산업도 많아집니다. 진단, 치료, 의료 서비스 개선 등 여러 방면에서 효과가 기대됩니다. 공공부문에서도 부정적인 시각은 거의 없다고 생각합니다. 물론 설득은 필요하겠지만, 긴 안목으로 볼 때 반드시 필요한 사업이라고 생각합니다. 차: 의료 데이터 활용 관련해서는 보안 문제도 자주 언급됩니다. 데이터 보안 문제를 해결할 방안도 갖고 계신가요? 김: 네, 아주 중요한 부분입니다. 최근 특정 기업에서 데이터 유출 문제가 발생했는데, 이는 원본 데이터를 보유하고 있었기 때문입니다. 원본 데이터가 있으면, 해킹이나 내부 유출의 위험이 항상 존재합니다. 은행도 마찬가지죠. 금고 안에 현금이 있기 때문에 도둑이 들어오는 것처럼, 원본 데이터를 가지고 있으면 다양한 방식으로 유출될 수 있습니다. 그러나 연합 학습(Federated Learning) 방식은 원본 데이터를 아예 수집하지 않습니다. 데이터는 병원이나 제약사 안에 그대로 있고, AI 모델이 그곳을 방문해 파라미터(가중치)만 받아오는 구조입니다. 이로 인해 원본 데이터 노출 위험이 현저히 낮아집니다. 또한, 이 구조에 다단계 암호화와 보안 계층을 추가하면, 해킹 위험을 더 줄일 수 있습니다. 우리가 일상적으로 사용하는 전자정보 서비스, 예를 들어 주민등록 등본 발급, 손택스, 민원24 같은 데서도 내 개인 정보가 유출됐다는 이야기를 들은 적이 거의 없잖아요. 그것은 정부가 강력한 관리 시스템을 갖추고 있기 때문입니다. 철저한 보안 관리 체계를 도입한다면 충분히 안전하게 운영될 수 있습니다. 차: 현재까지는 국민신약배당이 큰 그림 수준에서 논의된 것 같은데, 좀 더 구체적인 계획이나 전략이 있으실까요? 김: 기본 전략은 크게 세 가지입니다. 첫째는 ‘외국의 사례를 찾지 말자’는 겁니다. 외국이 해본 적이 있는지를 기준으로 삼으면 안 됩니다. 외국이 하지 않은 이유는 굳이 할 필요가 없었거나, 여건상 어렵기 때문입니다. 우리나라는 처음으로 이런 모델을 시도할 수 있는 나라이고, 최초의 생태계를 만들 수 있는 기회가 있습니다. 둘째는 ‘이해관계자 설득’입니다. 국민, 병원, 제약사, 공공기관, 정부 등 다양한 이해당사자들이 협의 테이블에 모여야 합니다. 이들이 하나의 공통된 목표를 위해 연대해야 가능성이 생깁니다. 셋째는 ‘지속 가능한 수익 모델’입니다. 어떤 사업이든 수익 구조가 있어야 지속 가능합니다. 비영리사업이라도 수익이 있어야 운영됩니다. 계속해서 정부 예산만 투입하는 방식으로는 장기적으로 유지하기 어렵습니다. 최근 다이어트 약이 연간 수십 조 원의 매출을 기록하고 있습니다. 이는 어떤 면에서는 국가 예산보다도 많을 정도입니다. 그만큼 바이오헬스 산업 시장은 계속 성장 중이고, 절대 줄지 않을 영역입니다. 유럽은 개인정보 이슈로 인해 이런 모델은 언급조차 어렵고, 미국은 거대 제약사들이 이미 자체적으로 발전할 수 있는 역량을 갖추고 있어 굳이 이런 방식이 필요 없습니다. 반면, 한국은 역설적으로 이런 방식이 가장 잘 맞는 나라입니다. 빠르게 기획하고 새로운 시스템을 만들 수 있는 능력이 있기 때문에, 오히려 유리한 입장에 있다고 생각합니다. 결국, 데이터를 거래하는 구조가 아니라, AI 모델이나 기능을 거래하는 구조로 가야 합니다. 이를 위해서는 여러 병원의 데이터를 활용할 수 있는 생태계가 필요하고요. 이 사업을 단순한 데이터 거래 사업이 아닌, AI 기반 서비스 산업 생태계로 이해해 주시면 좋겠습니다. 차: 네, 오늘은 K-MELLODDY 사업단 김화종 단장님 모시고 국민신약배당에 대한 이야기를 들어보았습니다. 단장님, 귀한 시간 내주셔서 감사합니다. 김: 감사합니다.2025-07-14 06:17:52김진구·차지현
"AI 신약개발 핵심, 데이터 확보...국민신약배당 필요"◆방송: DP초대석 ◆기획: 제약바이오산업2팀 김진구 기자 ◆진행: 제약바이오산업2팀 차지현 기자 ◆촬영·편집: 영상제작팀 ◆출연: 김화종 K-MELLODDY 사업단장 차지현(이하 차): 안녕하세요. 헬스케어 산업 내 다양한 인물을 만나 이야기를 나눠보는 DP 초대석 시간입니다. 오늘은 인공지능 기술과 바이오 산업의 융합을 통해 신약 개발의 새 패러다임을 열고 있는 K-MELLODDY 사업단의 김화종 단장님을 모셨습니다. 단장님, 안녕하세요. 김화종(이하 김): 안녕하세요. K-MELLODDY 사업단 단장을 맡고 있는 김화종입니다. 만나 뵙게 돼서 대단히 반갑습니다. 차: AI 기술이 매우 빠른 속도로 발달하고 있는데요. 최근 미국에서 직접 그 현장을 보고 듣고 오셨다고 들었습니다. 인상 깊었던 장면이 있는지 궁금합니다. 김: 지난 6월, 보스턴에서 열린 바이오USA에 참석했습니다. 130여 개 세션 중 20개 정도가 AI와 관련된 내용이었고요. 크게 세 가지로 나눌 수 있습니다. 첫째는 AI는 빠르게 발전하고 있지만, 바이오 분야에서는 발전이 더딘 편이라는 점입니다. 그 이유는 데이터가 부족하기 때문입니다. 바이오 관련 데이터는 개인 정보가 많고, 병원 데이터나 제약사의 실험 데이터는 외부에 공개되지 않고 있습니다. 둘째는 AI 활용에서 연결의 중요성이 강조됐다는 것입니다. 신약 개발에 AI를 활용할 때, 보통은 단계를 나눠서 활용합니다. 타깃을 찾고 검증하거나, 후보 물질을 찾고 검증하거나, 전임상 데이터를 쓰는 식이죠. 그런데 각 단계를 연결해 사용하는 게 더 효과적입니다. 궁극적으로 임상 1·2·3상을 통과하는 것이 목표이기 때문이죠. 여기에 최근에는 리얼월드 데이터를 AI 모델에 활용해야 한다는 논의도 많습니다. 실험은 매우 제한된 조건에서 이뤄지기 때문에, 실제 사람에게 적용했을 때 잘 맞지 않는 경우가 많습니다. 그래서 FDA에서도 동물실험을 줄이는 방향으로 가고 있고요. 처음부터 다양한 데이터를 함께 학습하는 AI 모델을 만들어야 한다는 얘기가 나왔습니다. 이런 이슈들이 바이오USA에서 핵심적으로 논의된 내용이었습니다. 차: AI 신약 개발 분야에서 K-MELLODDY 사업의 중요성도 점점 커지고 있는 것 같습니다. 이 사업은 어떤 사업이고, 출범한 지 1년 정도 지난 시점에서의 성과는 어떤지 궁금합니다. 김: K-MELLODDY 사업은 제가 한국제약바이오협회 AI신약개발지원센터장을 하던 2021년에 제안한 사업으로, 출범한 지는 4년 정도 됐습니다. 이 사업을 제안한 이유는, 빅데이터의 등장으로 AI 성능이 향상되고 있기 때문입니다. AI는 데이터가 많아야 잘 작동하는데, 신약 개발이나 바이오 분야는 데이터 확보가 굉장히 어렵습니다. 지금 우리가 많이 쓰는 인공지능은 언어모델이나 이미지 인식 분야입니다. 수억 개에서 수십억 개의 데이터를 학습했기 때문이죠. 반면 신약 개발 분야는 수백만 건도 안 되는 경우가 많습니다. 그래서 유럽에서는 10개 제약사가 모여 ‘멜로디(MELLODDY)’라는 프로젝트를 진행했습니다. 경쟁관계에 있지만 데이터를 공동 활용해서 더 좋은 성능의 AI 모델을 만들자는 거죠. 좋은 성능의 모델이 필요한 이유는 실패 확률을 줄이기 위해서입니다. K-MELLODDY는 이 유럽 사례를 참고해 시작됐지만, 구성은 다릅니다. 유럽은 제약사 중심이었다면, 우리는 병원·연구소·대학·창업사까지 모여 더 다양한 데이터를 확보하고자 했습니다. 사업비는 348억 원으로 적지는 않지만, 큰 성과를 내려면 부족한 수준이기도 해서 후속 사업을 고민 중입니다. 지난 1년간은 생태계를 조성하는 데 집중했습니다. 각 기관이 실험 조건과 방식을 공유했고, AI 모델 개발자와 데이터를 보유한 기관이 함께 협력해 모델을 개발하는 작업을 해왔습니다. 데이터가 외부로 나가지 않더라도, 데이터의 구조나 특성을 공유하면서 모델을 만들 수 있는 환경을 구축한 것이 주요 성과입니다. 차: 바이오 업계 사람들과 만나 보면, AI 신약 개발의 필요성은 공감하지만 아직 갈 길이 멀다는 이야기도 많이 합니다. AI 신약 개발에서 가장 중요한 것은 무엇이라고 보시나요? 김: 우리나라 개발자들이나 신약 개발자들의 시각과 글로벌 기업의 시각 차이는 상당히 큽니다. 글로벌은 세계 최고 수준의 인재와 자본이 모여 있기 때문에 첨단 기술이 나올 수밖에 없죠. 그렇다고 우리가 못할 일은 아닙니다. AI 개발에 필요한 기본적인 소프트웨어나 기술은 대부분 공개돼 있습니다. 출발선은 같다고 봐도 돼요. 누가 더 빠르게, 잘 활용하느냐가 관건입니다. 그런데 여기서 늘 문제가 되는 게 데이터입니다. 외국도 지금 데이터를 확보하려고 애쓰고 있고, 우리도 이 분야에 초점을 맞추면 충분히 경쟁력 있는 솔루션을 만들 수 있습니다. 결국 핵심은 ‘데이터 확보’이고, 우리나라도 이 부분은 충분히 할 수 있다고 봅니다. 차: 그렇다면 현재 한국의 AI 신약 개발 수준은 어느 정도라고 판단하시나요? 김: 출발선 자체는 큰 차이가 없습니다. 그런데 실제로는 차이가 큽니다. 예를 들어, AI 모델 정확도가 95%인 것과 96%인 것의 차이는 작아 보이지만, 사람들은 대부분 96%짜리를 선택합니다. 결국 1등 소프트웨어만 사용되는 거죠. 성능만 보면 비슷할 수 있어도, 실제 사용되는 데는 격차가 있습니다. 이걸 좁히기 위해서는 우리만의 무기가 필요합니다. 저는 그것이 ‘잘 정리된 한국의 의료 데이터’라고 생각합니다. 없는 게 아니라, 못 쓰고 있는 거죠. 이 데이터를 잘 활용하면 격차를 줄일 수 있고, 심지어 앞서갈 수도 있다고 생각합니다. 전 국민이 단일 건강보험 체계로 되어 있는 나라는 우리나라와 대만뿐입니다. 이 체계 덕분에 환자가 어떤 치료를 받았는지 쉽게 확인하고 연결할 수 있습니다. 이 데이터를 활용하면, 타깃 발굴, 바이오마커 검증, 약물 반응 추적까지 모두 가능해집니다. 차: 최근 ‘국민신약배당’이라는 새로운 개념을 제안하셨는데요. 어떤 사업인지, 또 어떻게 구상하게 되셨는지 궁금합니다. 김: 제가 ‘국민신약배당’이라는 이름의 사업을 제안했습니다. 이름만 보면 투자사업처럼 들릴 수도 있는데, 일부러 그런 이름을 붙였습니다. 우리나라만이 가진 강점이 바로 ‘잘 정리된 의료 데이터’입니다. 하지만 현재로선 제약이 많습니다. 그래서 이런 생태계를 만들기 위해서는 국민의 이해와 동의가 필요합니다. 지금까지 안 된 이유는 개인정보 보호 때문이라고 하지만, 사실은 개인이 자신의 데이터를 가지고 누군가가 이익을 얻는 것을 쉽게 허용하지 않기 때문이죠. 이 데이터가 공공재로 활용돼야 한다는 것이 제 생각입니다. 그리고 미래에 발생한 이익이 국민에게 배당되는 구조라면, 국민의 저항도 줄어들 겁니다. 좋은 데이터를 기반으로 블록버스터 신약이 만들어지면, 기업이 발전하고, 산업이 성장하고, 다시 국민에게 이익이 돌아가는 선순환이 만들어집니다. 이런 구조를 설명하고자 ‘국민신약배당’이라는 이름을 붙인 겁니다. 차: 국민신약배당 사업이 실제로 이뤄진다면, AI 신약 개발에 큰 도움이 될 것 같습니다. 이 사업을 진행하기 위해 가장 우선적으로 해결해야 할 문제는 무엇이라고 보시나요? 김: 방금 말씀드린 것처럼, 첫 번째는 국민의 동의입니다. 여러 각계각층의 사람들과 이야기해본 결과, 이런 사업이 있으면 좋겠다는 의견이 많았습니다. 산업이 발전하고, 우리나라가 주도하는 블록버스터 신약도 나올 수 있고, 나아가 배당이 이뤄져 건강보험료를 할인받거나 금전적으로 혜택을 얻을 수 있다면, 국민 입장에서도 안 받는 것보다는 낫다는 생각을 하게 될 겁니다. 이런 구조라면 국민 동의를 끌어낼 수 있다고 봅니다. 두 번째는 병원의 협조입니다. 현재는 병원에 가면, 환자들이 연구 목적의 데이터 활용을 대체로 허용하고 있습니다. 하지만 실제 연구는 매우 제한적입니다. IRB(기관생명윤리위원회) 승인을 받아야 하고, 병원 내 데이터만 활용 가능하며, 다른 병원 데이터는 사용할 수 없습니다. 그래서 병원에서도 특정 환자 데이터를 특정 목적에만 제한적으로 쓰던 상황에서, 여러 병원의 데이터를 폭넓게 활용하고, 다양한 AI 모델을 만들어 논문도 쓰고 연구도 할 수 있는 여건이 마련된다면, 오히려 연구 경쟁력을 키울 수 있습니다. 병원도 적극적으로 동참할 수 있는 기반이 생기는 것이죠. 차: 결국 모든 이해관계자들이 마음을 모아야 가능한 사업일 텐데요. 개인이나 병원뿐 아니라, 제약단체나 보건복지부 등 정부 차원의 공감대도 형성되어 있는지 궁금합니다. 김: 네, 사실 이 부분이 가장 중요합니다. 정부나 신약 개발 기업의 적극성이 핵심이죠. 제약기업들은 이런 사업을 환영할 것이라 봅니다. 데이터가 있는 것과 없는 것의 차이는 매우 큽니다. 데이터를 활용하면 신약을 설계하고, 만들고, 검증하는 데 큰 도움이 됩니다. 대신, 제약사들도 신약으로 큰 수익을 낸다면 일정 부분을 공공의 목적으로 환원해야 합니다. 정부 입장에서도 이 사업은 바이오산업을 키우는 중요한 기회입니다. 신약뿐 아니라, 이로 인해 파생되는 산업도 많아집니다. 진단, 치료, 의료 서비스 개선 등 여러 방면에서 효과가 기대됩니다. 공공부문에서도 부정적인 시각은 거의 없다고 생각합니다. 물론 설득은 필요하겠지만, 긴 안목으로 볼 때 반드시 필요한 사업이라고 생각합니다. 차: 의료 데이터 활용 관련해서는 보안 문제도 자주 언급됩니다. 데이터 보안 문제를 해결할 방안도 갖고 계신가요? 김: 네, 아주 중요한 부분입니다. 최근 특정 기업에서 데이터 유출 문제가 발생했는데, 이는 원본 데이터를 보유하고 있었기 때문입니다. 원본 데이터가 있으면, 해킹이나 내부 유출의 위험이 항상 존재합니다. 은행도 마찬가지죠. 금고 안에 현금이 있기 때문에 도둑이 들어오는 것처럼, 원본 데이터를 가지고 있으면 다양한 방식으로 유출될 수 있습니다. 그러나 연합 학습(Federated Learning) 방식은 원본 데이터를 아예 수집하지 않습니다. 데이터는 병원이나 제약사 안에 그대로 있고, AI 모델이 그곳을 방문해 파라미터(가중치)만 받아오는 구조입니다. 이로 인해 원본 데이터 노출 위험이 현저히 낮아집니다. 또한, 이 구조에 다단계 암호화와 보안 계층을 추가하면, 해킹 위험을 더 줄일 수 있습니다. 우리가 일상적으로 사용하는 전자정보 서비스, 예를 들어 주민등록 등본 발급, 손택스, 민원24 같은 데서도 내 개인 정보가 유출됐다는 이야기를 들은 적이 거의 없잖아요. 그것은 정부가 강력한 관리 시스템을 갖추고 있기 때문입니다. 철저한 보안 관리 체계를 도입한다면 충분히 안전하게 운영될 수 있습니다. 차: 현재까지는 국민신약배당이 큰 그림 수준에서 논의된 것 같은데, 좀 더 구체적인 계획이나 전략이 있으실까요? 김: 기본 전략은 크게 세 가지입니다. 첫째는 ‘외국의 사례를 찾지 말자’는 겁니다. 외국이 해본 적이 있는지를 기준으로 삼으면 안 됩니다. 외국이 하지 않은 이유는 굳이 할 필요가 없었거나, 여건상 어렵기 때문입니다. 우리나라는 처음으로 이런 모델을 시도할 수 있는 나라이고, 최초의 생태계를 만들 수 있는 기회가 있습니다. 둘째는 ‘이해관계자 설득’입니다. 국민, 병원, 제약사, 공공기관, 정부 등 다양한 이해당사자들이 협의 테이블에 모여야 합니다. 이들이 하나의 공통된 목표를 위해 연대해야 가능성이 생깁니다. 셋째는 ‘지속 가능한 수익 모델’입니다. 어떤 사업이든 수익 구조가 있어야 지속 가능합니다. 비영리사업이라도 수익이 있어야 운영됩니다. 계속해서 정부 예산만 투입하는 방식으로는 장기적으로 유지하기 어렵습니다. 최근 다이어트 약이 연간 수십 조 원의 매출을 기록하고 있습니다. 이는 어떤 면에서는 국가 예산보다도 많을 정도입니다. 그만큼 바이오헬스 산업 시장은 계속 성장 중이고, 절대 줄지 않을 영역입니다. 유럽은 개인정보 이슈로 인해 이런 모델은 언급조차 어렵고, 미국은 거대 제약사들이 이미 자체적으로 발전할 수 있는 역량을 갖추고 있어 굳이 이런 방식이 필요 없습니다. 반면, 한국은 역설적으로 이런 방식이 가장 잘 맞는 나라입니다. 빠르게 기획하고 새로운 시스템을 만들 수 있는 능력이 있기 때문에, 오히려 유리한 입장에 있다고 생각합니다. 결국, 데이터를 거래하는 구조가 아니라, AI 모델이나 기능을 거래하는 구조로 가야 합니다. 이를 위해서는 여러 병원의 데이터를 활용할 수 있는 생태계가 필요하고요. 이 사업을 단순한 데이터 거래 사업이 아닌, AI 기반 서비스 산업 생태계로 이해해 주시면 좋겠습니다. 차: 네, 오늘은 K-MELLODDY 사업단 김화종 단장님 모시고 국민신약배당에 대한 이야기를 들어보았습니다. 단장님, 귀한 시간 내주셔서 감사합니다. 김: 감사합니다.2025-07-14 06:17:52김진구·차지현 -
 식약처, CAR-T 이상반응 치료 '아나킨라' 긴급 도입 검토[데일리팜=이혜경 기자] 식품의약품안전처가 최근 CAR-T 세포치료제의 중증 이상반응 치료에 '아나킨라(Anakinra, 상품명 키너렛)'의 긴급도입 여부를 본격적으로 검토 중에 있는 것으로 알려졌다. 식약처는 지난 2일 전문지 출입기자단 공식 질의에 "현재 우리 처에서는 해당 품목 및 해당 적응증 관련 안전성, 유효성 및 해외 사용현황 등을 토대로 긴급도입의 필요성에 대하여 검토 중에 있다"고 밝혔다. 이는 대한혈액학회가 제출한 아나킨라 긴급도입 요청서를 기반으로 한 후속조치로, 민원을 접수한 의약품안전국이 관련 사안을 식약처 바이오의약품정책과로 이관해 본격적인 검토 절차에 들어간 것이다. CAR-T 세포 치료는 환자의 면역세포를 유전적으로 조작해 암세포를 공격하도록 하는 첨단 정밀치료법이다. 특히 재발성 및 난치성 급성 림프구성 백혈병, 미만성 거대 B세포 림프종, 다발골수종 등에서 기존 항암제와는 다른 효과를 기대할 수 있는 치료로 주목받는다. 국내에서는 치료 비용이 약 4~5억 원에 달하지만, 건강보험 급여가 적용되면서 환자 본인부담은 크게 줄어들었다. 그러나 이러한 고비용 치료가 급여 지원을 받는 반면, 치료 이후 발생할 수 있는 중증 면역 이상반응에 대응하는 일부 보조 약제는 여전히 제도권 밖에 머무르고 있다. 대부분 '토실리주맙'이나 '고용량 스테로이드'가 1차 약제로 쓰이지만, 이에 불응하는 환자에 대해서는 인터루킨-1(IL-1) 억제제인 '아나킨라'가 중요한 치료 대안으로 활용되고 있다. 하지만 아나킨라는 제조사 판단에 의해 국내에서 철수된 이후 일반적인 사용이 막혀 있는 상태다. 아나킨라는 스웨덴의 소비(Sobi)사에서 제조한 인터루킨-1 수용체 길항제로, 미국(FDA), 유럽(EMA), 일본(PMDA) 등에서는 류마티스관절염, CAPS, FMF, Still's Disease 등에 대한 치료제로 승인되어 있다. 2025년판 GoCART Coalition의 유럽 CAR-T Handbook에서는 CRS 및 ICANS 발생 시 고용량 아나킨라 투여를 권고하고 있으며, IEC-HS 환자에 대해서는 코르티코스테로이드와 병용 투여가 지침에 포함돼 있다. 특히 스테로이드와 토실리주맙 치료에도 반응하지 않는 고위험 환자에게는 필수 치료 옵션으로 제시되고 있다. 또한 Gazeau N 외 연구(Transplant Cell Ther. 2023)에서는 CAR-T 치료 후 Grade 2 이상 합병증이 발생한 43명의 환자에게 아나킨라를 투여한 결과, 28일 내 치료 관련 사망률은 0%, 전체 반응률은 77%로 나타났다. 반면, 국내에서는 아나킨라가 긴급도입의약품으로 한국희귀·필수의약품센터를 통해 제한적으로 공급되고 있지만, CAR-T 이상반응 치료에는 사용이 불가능하다. 이에 일부 병원에서는 자체 IRB 승인을 통해 제한적 처방을 시도하고 있지만, 보험급여나 법적 보호를 받을 수 없어 전국 단위의 적용은 불가능한 상황이다. 대한혈액학회는 "현재 의료진들은 환자의 생명을 살리기 위해, 불법 진료의 위험을 감수하고라도 비공식적으로 아나킨라를 사용할 수밖에 없는 상황에 처해 있다"며 "이는 국가 공공 보건제도 및 생명권 보장의 기본 원칙에 부합하지 않으며, 빠른 시일 내 제도적 정비가 절실하다"고 밝혔다. CAR-T 치료제의 급여 적용 이후 수년간 복지부와 심평원 등에 민원을 제기해온 대한혈액학회는 지난 4월 국민신문고를 통해 아나킨라의 적응증 외 사용 허용 및 제도 개선에 대한 민원을 제출했으며, 이에 대해 식약처는 "중앙행정기관 또는 전문단체의 요청 시, 환자치료상 필수성, 대체가능성, 해외 허가 현황 등을 바탕으로 인정범위 확대를 검토할 수 있다"고 회신했다. 이에 혈액학회는 5월 19일 식약처 의약품안전국에 아나킨라 긴급도입 요청서를 정식 제출했다. 요청서에는 품목정보, 해외 주요국 허가 현황, 미국 및 유럽에서의 사용 사례, 환자 생명을 위협하는 상황에서의 사용 필요성, 국내 수요 예측(연간 100명 미만), 약물 대체 가능성 부족 등의 내용을 포함했다. 대한혈액학회는 "생명을 위협하는 중증 합병증에서의 빠른 면역 조절이 반드시 필요하며, 현재 국내에는 아나킨라를 대체할 만한 유사 기전의 약물이 존재하지 않는다"며 "이번 긴급도입 요청은 환자 생명을 보호하고 치료 접근성을 보장하기 위한 필수적 조치로서, 조속한 제도 정비가 요구된다"고 강조했다.2025-07-02 17:04:27이혜경
식약처, CAR-T 이상반응 치료 '아나킨라' 긴급 도입 검토[데일리팜=이혜경 기자] 식품의약품안전처가 최근 CAR-T 세포치료제의 중증 이상반응 치료에 '아나킨라(Anakinra, 상품명 키너렛)'의 긴급도입 여부를 본격적으로 검토 중에 있는 것으로 알려졌다. 식약처는 지난 2일 전문지 출입기자단 공식 질의에 "현재 우리 처에서는 해당 품목 및 해당 적응증 관련 안전성, 유효성 및 해외 사용현황 등을 토대로 긴급도입의 필요성에 대하여 검토 중에 있다"고 밝혔다. 이는 대한혈액학회가 제출한 아나킨라 긴급도입 요청서를 기반으로 한 후속조치로, 민원을 접수한 의약품안전국이 관련 사안을 식약처 바이오의약품정책과로 이관해 본격적인 검토 절차에 들어간 것이다. CAR-T 세포 치료는 환자의 면역세포를 유전적으로 조작해 암세포를 공격하도록 하는 첨단 정밀치료법이다. 특히 재발성 및 난치성 급성 림프구성 백혈병, 미만성 거대 B세포 림프종, 다발골수종 등에서 기존 항암제와는 다른 효과를 기대할 수 있는 치료로 주목받는다. 국내에서는 치료 비용이 약 4~5억 원에 달하지만, 건강보험 급여가 적용되면서 환자 본인부담은 크게 줄어들었다. 그러나 이러한 고비용 치료가 급여 지원을 받는 반면, 치료 이후 발생할 수 있는 중증 면역 이상반응에 대응하는 일부 보조 약제는 여전히 제도권 밖에 머무르고 있다. 대부분 '토실리주맙'이나 '고용량 스테로이드'가 1차 약제로 쓰이지만, 이에 불응하는 환자에 대해서는 인터루킨-1(IL-1) 억제제인 '아나킨라'가 중요한 치료 대안으로 활용되고 있다. 하지만 아나킨라는 제조사 판단에 의해 국내에서 철수된 이후 일반적인 사용이 막혀 있는 상태다. 아나킨라는 스웨덴의 소비(Sobi)사에서 제조한 인터루킨-1 수용체 길항제로, 미국(FDA), 유럽(EMA), 일본(PMDA) 등에서는 류마티스관절염, CAPS, FMF, Still's Disease 등에 대한 치료제로 승인되어 있다. 2025년판 GoCART Coalition의 유럽 CAR-T Handbook에서는 CRS 및 ICANS 발생 시 고용량 아나킨라 투여를 권고하고 있으며, IEC-HS 환자에 대해서는 코르티코스테로이드와 병용 투여가 지침에 포함돼 있다. 특히 스테로이드와 토실리주맙 치료에도 반응하지 않는 고위험 환자에게는 필수 치료 옵션으로 제시되고 있다. 또한 Gazeau N 외 연구(Transplant Cell Ther. 2023)에서는 CAR-T 치료 후 Grade 2 이상 합병증이 발생한 43명의 환자에게 아나킨라를 투여한 결과, 28일 내 치료 관련 사망률은 0%, 전체 반응률은 77%로 나타났다. 반면, 국내에서는 아나킨라가 긴급도입의약품으로 한국희귀·필수의약품센터를 통해 제한적으로 공급되고 있지만, CAR-T 이상반응 치료에는 사용이 불가능하다. 이에 일부 병원에서는 자체 IRB 승인을 통해 제한적 처방을 시도하고 있지만, 보험급여나 법적 보호를 받을 수 없어 전국 단위의 적용은 불가능한 상황이다. 대한혈액학회는 "현재 의료진들은 환자의 생명을 살리기 위해, 불법 진료의 위험을 감수하고라도 비공식적으로 아나킨라를 사용할 수밖에 없는 상황에 처해 있다"며 "이는 국가 공공 보건제도 및 생명권 보장의 기본 원칙에 부합하지 않으며, 빠른 시일 내 제도적 정비가 절실하다"고 밝혔다. CAR-T 치료제의 급여 적용 이후 수년간 복지부와 심평원 등에 민원을 제기해온 대한혈액학회는 지난 4월 국민신문고를 통해 아나킨라의 적응증 외 사용 허용 및 제도 개선에 대한 민원을 제출했으며, 이에 대해 식약처는 "중앙행정기관 또는 전문단체의 요청 시, 환자치료상 필수성, 대체가능성, 해외 허가 현황 등을 바탕으로 인정범위 확대를 검토할 수 있다"고 회신했다. 이에 혈액학회는 5월 19일 식약처 의약품안전국에 아나킨라 긴급도입 요청서를 정식 제출했다. 요청서에는 품목정보, 해외 주요국 허가 현황, 미국 및 유럽에서의 사용 사례, 환자 생명을 위협하는 상황에서의 사용 필요성, 국내 수요 예측(연간 100명 미만), 약물 대체 가능성 부족 등의 내용을 포함했다. 대한혈액학회는 "생명을 위협하는 중증 합병증에서의 빠른 면역 조절이 반드시 필요하며, 현재 국내에는 아나킨라를 대체할 만한 유사 기전의 약물이 존재하지 않는다"며 "이번 긴급도입 요청은 환자 생명을 보호하고 치료 접근성을 보장하기 위한 필수적 조치로서, 조속한 제도 정비가 요구된다"고 강조했다.2025-07-02 17:04:27이혜경 -
 KCDA-KAIRB, 임상시험 환경 조성 업무협약 체결[데일리팜=황병우 기자] 한국임상개발협회(회장 임윤희, 이하 KCDA)는 국내 임상시험 산업의 발전과 효율적인 연구 환경 조성을 위해 대한기관윤리심의기구협의회(회장 김병수, 이하 KAIRB)와 업무협약(MOU)을 체결했다고 25일 밝혔다. 이번 업무협약에 따라 양 기관은 임상 분야의 최신 동향을 반영한 정책 개발 관련 논의와 공동 연구를 통해 국내 임상시험 산업의 발전과 효율적인 연구 환경, 국제적 경쟁력 강화를 위해 긴밀히 협력할 계획이다. 구체적으로 임상 분야에 대한 ▲임상시험 환경 및 제도 개선, 효율성 증대를 위한 논의 및 협력 ▲최신 동향 정보 교류 ▲정책 수립 지원 및 정책 공동 연구 ▲포럼 & 8231; 세미나 등 행사 개최 협력 등을 통해 상호 협력한다. 한국임상개발협회(KCDA)는 임상시험의 전 주기를 아우르며, 임상시험 종사자의 전문성 향상과 국내 신약개발 역량 강화를 목표로 학술·연구·교육 활동을 수행하는 임상시험 분야의 대표적인 비영리 사단법인이다. 현재 협회에는 제약사, 바이오벤처, 의료기관, 임상시험수탁기관(CRO), 솔루션 기업 등 110여 개의 회원사가 가입되어 있다. 주요 사업으로 ▲임상시험 종사자 대상 교육 프로그램 운영 ▲글로벌 동향 기반의 분과 세미나 및 전략 연구 ▲정부 및 규제기관과의 정책 협의 ▲유관 단체와의 네트워크 협력 등이 있다. 또 대한기관윤리심의기구협의회(KAIRB)는 국내 유일의 기관생명윤리위원회(IRB) 비영리 법인으로 2007년에 설립돼 임상시험을 포함한 인간 대상 연구가 과학적이고 윤리적으로 수행되기 위한 여건을 마련 중이다. 임윤희 한국임상개발협회장은 "KAIRB와의 업무협약을 통해 국내 임상개발 생태계의 신뢰성과 전문성을 강화하고 효율적인 운영을 위한 중요한 협력 기반을 마련하게 되어 매우 뜻깊다"고 말했다. 이어 김병수 대한기관윤리심의기구협의회 회장은 "우리나라 임상시험 경쟁력 강화를 위해 한국임상개발협회와 협력 체계를 구축할 수 있게 되어 기쁘다"며 "협의회의 설립 취지에 따라 윤리적이고도 과학적인 임상시험 환경 조정 및 국제적 경쟁력 강화를 위해 적극적이면서도 지속적으로 노력할 것"이라고 덧붙였다.2025-04-25 11:58:28황병우
KCDA-KAIRB, 임상시험 환경 조성 업무협약 체결[데일리팜=황병우 기자] 한국임상개발협회(회장 임윤희, 이하 KCDA)는 국내 임상시험 산업의 발전과 효율적인 연구 환경 조성을 위해 대한기관윤리심의기구협의회(회장 김병수, 이하 KAIRB)와 업무협약(MOU)을 체결했다고 25일 밝혔다. 이번 업무협약에 따라 양 기관은 임상 분야의 최신 동향을 반영한 정책 개발 관련 논의와 공동 연구를 통해 국내 임상시험 산업의 발전과 효율적인 연구 환경, 국제적 경쟁력 강화를 위해 긴밀히 협력할 계획이다. 구체적으로 임상 분야에 대한 ▲임상시험 환경 및 제도 개선, 효율성 증대를 위한 논의 및 협력 ▲최신 동향 정보 교류 ▲정책 수립 지원 및 정책 공동 연구 ▲포럼 & 8231; 세미나 등 행사 개최 협력 등을 통해 상호 협력한다. 한국임상개발협회(KCDA)는 임상시험의 전 주기를 아우르며, 임상시험 종사자의 전문성 향상과 국내 신약개발 역량 강화를 목표로 학술·연구·교육 활동을 수행하는 임상시험 분야의 대표적인 비영리 사단법인이다. 현재 협회에는 제약사, 바이오벤처, 의료기관, 임상시험수탁기관(CRO), 솔루션 기업 등 110여 개의 회원사가 가입되어 있다. 주요 사업으로 ▲임상시험 종사자 대상 교육 프로그램 운영 ▲글로벌 동향 기반의 분과 세미나 및 전략 연구 ▲정부 및 규제기관과의 정책 협의 ▲유관 단체와의 네트워크 협력 등이 있다. 또 대한기관윤리심의기구협의회(KAIRB)는 국내 유일의 기관생명윤리위원회(IRB) 비영리 법인으로 2007년에 설립돼 임상시험을 포함한 인간 대상 연구가 과학적이고 윤리적으로 수행되기 위한 여건을 마련 중이다. 임윤희 한국임상개발협회장은 "KAIRB와의 업무협약을 통해 국내 임상개발 생태계의 신뢰성과 전문성을 강화하고 효율적인 운영을 위한 중요한 협력 기반을 마련하게 되어 매우 뜻깊다"고 말했다. 이어 김병수 대한기관윤리심의기구협의회 회장은 "우리나라 임상시험 경쟁력 강화를 위해 한국임상개발협회와 협력 체계를 구축할 수 있게 되어 기쁘다"며 "협의회의 설립 취지에 따라 윤리적이고도 과학적인 임상시험 환경 조정 및 국제적 경쟁력 강화를 위해 적극적이면서도 지속적으로 노력할 것"이라고 덧붙였다.2025-04-25 11:58:28황병우 -
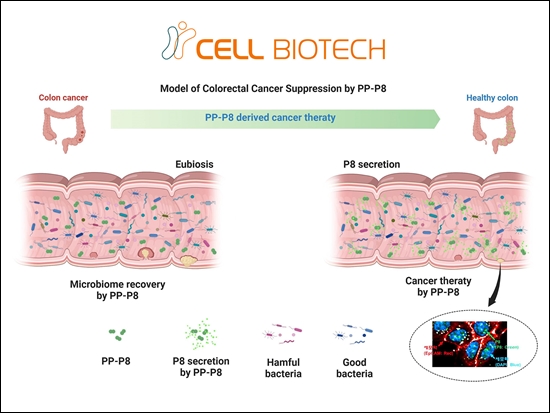 쎌바이오텍, 대장암신약 'PP-P8' 임상시험 개시[데일리팜=노병철 기자] K-유산균 대장암 치료 혁신 ‘듀오락(DUOLAC)’을 전개하는 쎌바이오텍이 마이크로바이옴을 활용한 대장암 신약 ‘PP-P8’의 임상시험을 시작하며, 바이오의약품 기업으로 본격 도약한다. 쎌바이오텍은서울대학교병원과 함께‘PP-P8’의 첫 환자 투약을 시작하며,임상시험을 본격 개시한다고 19일 밝혔다. 쎌바이오텍은 지난해 3월, 국내 식품의약품안전처로부터 ‘PP-P8’의 1상 임상시험계획(IND)을 승인받았다. 이후 서울대학교병원과 협력해환자 선정기준을 구체화하고, 병용금기약물을 조정하는 등 프로토콜을 변경했다. 변경된 임상시험계획은 서울대학교병원 의학연구윤리심의위원회(IRB)와 식품의약품안전처의 승인을 거쳐,본격적인 환자 투약이 시작되었다. 예상보다 시간이 소요되었으나, 이는 환자의 안전을 최우선으로 고려한 철저한 준비와 검토 과정이었다고 회사 측은 설명했다. 임상시험은 서울대학교병원에서 총 32명의 전이성 대장암(직결장암) 환자를 대상으로 ▲내약성 ▲안전성 ▲유효성을 평가한다. 용량 증량 단계(Part 1)에서는 단계적으로 투약 용량을 증량하여 안전성을 확인하고, 용량 확장 단계(Part 2)에서는 적정 용량을 선정해 유효성을 탐색한다. PP-P8은 유산균 유전자를 재조합해 항암제로 개발하는 국내 최초의 혁신 신약(First-in-Class)이다. 구체적으로 듀오락의 특허 균주인 ‘CBT-LR5(Lactobacillus Rhamnosus CBT-LR5, KCTC 12202BP)’에서 유래한 항암 단백질 ‘P8’을 대량 복제 생산하도록 개발된 ‘CBT-SL4(PediococcusPentosaceus CBT-SL4, KCTC 10297BP)’ 기반의 형질전환 유산균이다. 이 기술은 혁신적인 유전자 조작 기술을 활용해 대장암 세포를 사멸시키는 항암 단백질 ‘P8’을 자연 상태보다 100배 이상 생산할 수 있는 메커니즘으로 설계됐다. 임상시험에 필요한 모든 시약은 쎌바이오텍의 김포 본사 내 ‘생물학적 제제 의약품 공장’에서 직접 생산된다. 현재 전 세계적으로 미생물을 활용한 마이크로바이옴 신약 개발이 활발히 이루어지고 있지만, 신약 및 임상용 시약을 생산할 수 있는 CDMO(위탁개발생산) 기업은 극히 드문 실정이다. 쎌바이오텍은 지난 30년간 듀오락 사업을 통해 축적한 유산균 발효 기술과 노하우를 기반으로 마이크로바이옴 관련 의약품 균주의 글로벌 CDMO 시장을 선도하는 것을 목표로 한다. 쎌바이오텍 변종선 임상개발팀장은 “유산균 약물전달시스템(DDS) 플랫폼 기술을 활용해 대장암 신약뿐만 아니라 당뇨와 비만, 질염 등 다양한 질환에 대한 신약 개발로 사업 영역을 확장하고 있다”며, “쎌바이오텍은 축적된 노하우와 경험을 바탕으로 신약 개발 기간을 단축하고 시행착오를 줄이며 바이오의약품 기업으로 거듭날 것”이라고 전했다. 한편, CBT유산균으로 ‘K-유산균’ 대장암 치료 혁신을 이끄는 쎌바이오텍은 11년 연속 세계 수출 1위를 기록하는데 이어, 글로벌 유산균 시장에서의 입지와 경쟁력을 인정받아 ‘2024 세계일류상품’으로 선정됐다. 쎌바이오텍은 유산균 브랜드 듀오락을 앞세워 현재 전 세계 55개국에 CBT유산균을 수출하고 있으며, 유산균 본고장 덴마크에서 시장점유율 2위를 기록하고 있다.2025-02-19 09:58:34노병철
쎌바이오텍, 대장암신약 'PP-P8' 임상시험 개시[데일리팜=노병철 기자] K-유산균 대장암 치료 혁신 ‘듀오락(DUOLAC)’을 전개하는 쎌바이오텍이 마이크로바이옴을 활용한 대장암 신약 ‘PP-P8’의 임상시험을 시작하며, 바이오의약품 기업으로 본격 도약한다. 쎌바이오텍은서울대학교병원과 함께‘PP-P8’의 첫 환자 투약을 시작하며,임상시험을 본격 개시한다고 19일 밝혔다. 쎌바이오텍은 지난해 3월, 국내 식품의약품안전처로부터 ‘PP-P8’의 1상 임상시험계획(IND)을 승인받았다. 이후 서울대학교병원과 협력해환자 선정기준을 구체화하고, 병용금기약물을 조정하는 등 프로토콜을 변경했다. 변경된 임상시험계획은 서울대학교병원 의학연구윤리심의위원회(IRB)와 식품의약품안전처의 승인을 거쳐,본격적인 환자 투약이 시작되었다. 예상보다 시간이 소요되었으나, 이는 환자의 안전을 최우선으로 고려한 철저한 준비와 검토 과정이었다고 회사 측은 설명했다. 임상시험은 서울대학교병원에서 총 32명의 전이성 대장암(직결장암) 환자를 대상으로 ▲내약성 ▲안전성 ▲유효성을 평가한다. 용량 증량 단계(Part 1)에서는 단계적으로 투약 용량을 증량하여 안전성을 확인하고, 용량 확장 단계(Part 2)에서는 적정 용량을 선정해 유효성을 탐색한다. PP-P8은 유산균 유전자를 재조합해 항암제로 개발하는 국내 최초의 혁신 신약(First-in-Class)이다. 구체적으로 듀오락의 특허 균주인 ‘CBT-LR5(Lactobacillus Rhamnosus CBT-LR5, KCTC 12202BP)’에서 유래한 항암 단백질 ‘P8’을 대량 복제 생산하도록 개발된 ‘CBT-SL4(PediococcusPentosaceus CBT-SL4, KCTC 10297BP)’ 기반의 형질전환 유산균이다. 이 기술은 혁신적인 유전자 조작 기술을 활용해 대장암 세포를 사멸시키는 항암 단백질 ‘P8’을 자연 상태보다 100배 이상 생산할 수 있는 메커니즘으로 설계됐다. 임상시험에 필요한 모든 시약은 쎌바이오텍의 김포 본사 내 ‘생물학적 제제 의약품 공장’에서 직접 생산된다. 현재 전 세계적으로 미생물을 활용한 마이크로바이옴 신약 개발이 활발히 이루어지고 있지만, 신약 및 임상용 시약을 생산할 수 있는 CDMO(위탁개발생산) 기업은 극히 드문 실정이다. 쎌바이오텍은 지난 30년간 듀오락 사업을 통해 축적한 유산균 발효 기술과 노하우를 기반으로 마이크로바이옴 관련 의약품 균주의 글로벌 CDMO 시장을 선도하는 것을 목표로 한다. 쎌바이오텍 변종선 임상개발팀장은 “유산균 약물전달시스템(DDS) 플랫폼 기술을 활용해 대장암 신약뿐만 아니라 당뇨와 비만, 질염 등 다양한 질환에 대한 신약 개발로 사업 영역을 확장하고 있다”며, “쎌바이오텍은 축적된 노하우와 경험을 바탕으로 신약 개발 기간을 단축하고 시행착오를 줄이며 바이오의약품 기업으로 거듭날 것”이라고 전했다. 한편, CBT유산균으로 ‘K-유산균’ 대장암 치료 혁신을 이끄는 쎌바이오텍은 11년 연속 세계 수출 1위를 기록하는데 이어, 글로벌 유산균 시장에서의 입지와 경쟁력을 인정받아 ‘2024 세계일류상품’으로 선정됐다. 쎌바이오텍은 유산균 브랜드 듀오락을 앞세워 현재 전 세계 55개국에 CBT유산균을 수출하고 있으며, 유산균 본고장 덴마크에서 시장점유율 2위를 기록하고 있다.2025-02-19 09:58:34노병철 -
![[칼럼] 자연 재해와 임상시험](https://pds.dailypharm.com/news_image/202412/318714_1.jpg) [칼럼] 자연 재해와 임상시험임상시험은 취약하다. 데이터 조작, 환자 조작 등이 임상시험 중에 발생하기도 한다. MERS, Covid-19 등 공중보건 위기 상황에서는 임상시험 참여자가 병원에 방문할 수 없고, 스폰서 또는 CRO 임상책임자들의 병원 출입이 제한되기도 한다. 임상시험은 각종 재해(災害)로 중단되거나 수집된 데이터가 심각하게 훼손되기도 한다. 필자는 임상시험 중 일어나는 자연재해에 대해 말하고자 한다. 미국 국립암연구소(US National Cancer Institute)에서 일하고 있던 1980년, 미국 국립 신경 질환 및 뇌졸중 연구소(US National Institute of Neurological Disorder and Stroke, NINDS)의 전직 요청을 받고 자리를 옮겼다. NINDS에서 중요한 임상을 계획 중이었고 임상시험 전문가인 필자를 필요로 했다. 영유아들이 독감에 걸리면 열이 나면서 경련(seizure)을 일으키는 경우가 있다. 경련이 반복되거나 악성일 경우, 19세기부터 페노바르비탈(phenobarbital, 수면제 진정제의 일종)이라는 항경련제(anti-seizure drug)를 처방해 왔다. 독감으로 열성 경련(febrile seizure)을 앓는 영유아들이 3~4%가 되기 때문에 심각한 문제였지만 동시에 페노바르비탈을 영유아에게 처방하는 것이 안전하냐는 의문도 꾸준히 제기되고 있었다. 필자는 이와 관련된 임상시험을 담당했고, 2년 이상의 사전 연구를 거쳐 임상시험이 시작되었다. 1982년 11월 시작된 임상시험은 7년이 걸려 1989년에 마무리됐고 1990년 2월, 세계 최고 권위 의학 학술지 뉴잉글랜드 저널 오브 메디슨(New England Journal of Medicine)에 게재되었다. 임상시험 결과는 뉴욕타임즈(The New York Times)에 1면 top으로 소개되었고 당시 미국 의료계를 뒤집는 사건이었다. 페노바르비탈은 열성 경련에 아무런 효과가 없을 뿐 아니라, 이를 장기간 복용한 영유아는 지능지수(IQ score)가 심각하게 떨어진다는 결론이었다. 이렇게 중요한 페노바르비탈 임상시험 뒤에는 비화가 있다. 이 임상시험을 날려버릴 뻔한 자연재해가 있었다. 당시 임상시험 데이터를 사이트(site)에서 전화선으로, 휴렛팩커드(Hewlett-Packard, HP)에서 생산한 마이크로컴퓨터(micro-computer)에 직접 올리는 원격 데이터 입력 시스템(remote data entry system)이 처음 시작되었다. 필자는 페이퍼 데이터 관리(paper data management)를 주장했으나 필자의 의견보다는 컴퓨터 책임자들의 의견이 우세했다. 필자는 아직 검증되지 않은 IT 기술에 중대한 임상시험을 맡길 수 없다는 입장이었고 컴퓨터 전문가들은 HP를 못 믿으면 누구를 믿겠냐는 입장이었다. HP는 당시 세계 최고의 전자기기 회사였다. 결국 필자도 사이트에서 페이퍼로 데이터를 백업하는 조건으로 합의했다. 약 2년간 잘 진행되었고 필자도 할 말이 없었다. 필자의 관여에 대해 쓸데없는 짓이라는 힐난이 있었지만 데이터는 백업 되어야 한다는 입장을 고수했다. 1984년 여름으로 기억한다. 워싱턴 D.C.에 기록적 폭풍과 폭우가 닥쳤다. 천둥 번개가 밤새 계속되었고 정전사태가 벌어졌다. 다음 날 아침, 출근하자마자 확인해 보니 HP 마이크로 컴퓨터 디스크(micro computer disc)가 백업 디스크(back-up disc)와 더불어 모두 깨져 있었다. 그 동안 수집된 데이터가 모두 날아간 것이다. 밤새 친 천둥번개로 인한 파워 서지(power surge)가 원인이라고 컴퓨터 담당자가 설명했다. 연구소(Institute)에서는 난리가 났다. 필자는 페이퍼로 사이트에 백업이 되어 있으니 걱정하지 말라고 주변을 안심시키고, 페이퍼 데이터 관리를 다시 시작함으로써 임상시험을 성공리에 마칠 수 있었다. 필자는 이 임상시험을 성공리에 마쳐서 연구소(Institute)에서 표창을 받고 미국 국립 아동 건강 및 인간 발달 연구소(National Institute of Child Health and Human Development)에서 Biometry Chief를 제의 받고 자리를 옮겼다. 이번에는 4500명을 대상으로 하는 임신중독증 관련 임상시험이 기다리고 있었다. 임상시험이 자연재해에 취약함을 필자는 몸소 경험했다. 미국에서는 인터넷이 개발되면서 1990년대 중반부터 EDC(Electronic Data Capture)가 도입되었지만 2000년도 초기만 해도 페이퍼 백업(paper backup)을 요구하였다. 이제는 페이퍼 백업은 모두 사라졌다. 그러나 자연재해에 의한 임상시험의 취약점이 사라진 것은 아니다. 임상시험은 전 세계에서 진행되고 있다. 국내 바이오-제약사들의 많은 임상시험 역시 전 세계에서 진행되고 있다. 임상시험에 미칠 자연재해가 남의 일이 아니다. 자연재해 뿐 아니다. 인재(人災) 역시 심각한 문제다. 2018년 11월 KT 아현지사의 화재 사건, 2022년 10월 카카오 데이터 센터의 화재 사건 등은 전형적 인재다. 이런 인재도 임상시험을 파괴할 수 있다. 자연재해의 몇 가지 사례를 보자. 1990년 9월 수해로 아산병원이 침수되었다. 2001년 6월 미국 텍사스 대학교 건강과학센터(University of Texas Health Science Center)의 임상연구 시설과 IRB 사무실이 열대성 폭풍 앨리슨(Allison)으로 침수되었다. 2005년 뉴올리언즈 옥스너 의료센터(Ochsner Medical Center)에서 허리케인 카트리나(Katrina)로 인해 항암 임상시험 중인 입원 환자들이 미국내 여러 시설로 긴급 철수해야 했다. 2011년 뉴질랜드 크라이스트처치(Christchruch) 지진으로 병원이 파괴되고 연속되는 여진으로 몇 개월간 병원에 접근을 할 수 없었다. 푸에르토리코는 거의 상시로 발생하는 허리케인 위협으로 인해 임상시험 계획에 비상 대책을 포함한다. 우리나라도 아산병원 침수사건 이래 행정안전부는 지하공간 침수방지를 위한 수방기준 실무매뉴얼을 발행했다. 허리케인 카트리나를 겪은 루이지애나 주립대학교(Louisiana State University) 연구자들은 자연재해에 대비하여 두 가지를 강조했다. 첫째는 임상시험에 참여하는 환자와 커뮤니케이션 플랜을 준비할 것을 강조했다. 둘째는 사이트에 닥칠 재해에 대비해 리서치 데이터의 안전성 대책을 세울 것을 강조했다. 마이애미 대학교(University of Miami)에서는 허리케인대비 임상연구 매뉴얼을 발행했다. 연구자가 준비해야 할 사항들, 임상연구 참여자들을 위한 고려사항, 임상시험 중 수집되는 샘플 보호 등에 대한 비상 대책, 데이터 보호대책, 자연재해 후에 임상연구 연속성에 대한 대책 등으로 구성되어 있다. 미국 FDA는 2023년 9월 재난(Disasters)과 공중건강위기(Public Health Emergencies)로 인한 혼란상황에서의 임상시험 진행에 관한 지침(guidance)을 발행했다. FDA 지침의 목적은 위기와 혼란상황에서 임상참여자들을 보호하고, 임상시험 관리기준(GCP)를 유지하고, 임상시험에 미칠 위험을 최소화할 수 있도록 스폰서, IRB, 연구자들을 지원하는 데 있다고 했다. 그리고 Q&A에서 22개의 상황을 상정해 대응 방안에 대하여 논의하고 있다. 임상시험 데이터는 자연재해 또는 인재로부터 보호받기 위한 대책이 철저하게 준비되어야 한다. 2022년 태풍 '힌남노'로 포항제철은 135일간 생산공정이 중단되었고 그 손해가 2조가 넘었다고 한다. 우리는 해마다 태풍, 폭설 등 자연재해와 끊임없는 화재로 많은 피해를 입고 있다. 임상시험도 자연재해와 인재에 취약하다는 사실을 염두에 두고 대비책을 세우는 것이 중요할 것 같다. 더욱이 국내 제약 바이오사들도 MRCTs(Multi-Regional Clinical Trials, 다지역·다국가 임상시험)를 많이 진행하고 있다. 재해는 시간과 장소를 가리지 않는다. 각종 재해를 대비해야 할 것이다.2024-12-16 08:50:19데일리팜
[칼럼] 자연 재해와 임상시험임상시험은 취약하다. 데이터 조작, 환자 조작 등이 임상시험 중에 발생하기도 한다. MERS, Covid-19 등 공중보건 위기 상황에서는 임상시험 참여자가 병원에 방문할 수 없고, 스폰서 또는 CRO 임상책임자들의 병원 출입이 제한되기도 한다. 임상시험은 각종 재해(災害)로 중단되거나 수집된 데이터가 심각하게 훼손되기도 한다. 필자는 임상시험 중 일어나는 자연재해에 대해 말하고자 한다. 미국 국립암연구소(US National Cancer Institute)에서 일하고 있던 1980년, 미국 국립 신경 질환 및 뇌졸중 연구소(US National Institute of Neurological Disorder and Stroke, NINDS)의 전직 요청을 받고 자리를 옮겼다. NINDS에서 중요한 임상을 계획 중이었고 임상시험 전문가인 필자를 필요로 했다. 영유아들이 독감에 걸리면 열이 나면서 경련(seizure)을 일으키는 경우가 있다. 경련이 반복되거나 악성일 경우, 19세기부터 페노바르비탈(phenobarbital, 수면제 진정제의 일종)이라는 항경련제(anti-seizure drug)를 처방해 왔다. 독감으로 열성 경련(febrile seizure)을 앓는 영유아들이 3~4%가 되기 때문에 심각한 문제였지만 동시에 페노바르비탈을 영유아에게 처방하는 것이 안전하냐는 의문도 꾸준히 제기되고 있었다. 필자는 이와 관련된 임상시험을 담당했고, 2년 이상의 사전 연구를 거쳐 임상시험이 시작되었다. 1982년 11월 시작된 임상시험은 7년이 걸려 1989년에 마무리됐고 1990년 2월, 세계 최고 권위 의학 학술지 뉴잉글랜드 저널 오브 메디슨(New England Journal of Medicine)에 게재되었다. 임상시험 결과는 뉴욕타임즈(The New York Times)에 1면 top으로 소개되었고 당시 미국 의료계를 뒤집는 사건이었다. 페노바르비탈은 열성 경련에 아무런 효과가 없을 뿐 아니라, 이를 장기간 복용한 영유아는 지능지수(IQ score)가 심각하게 떨어진다는 결론이었다. 이렇게 중요한 페노바르비탈 임상시험 뒤에는 비화가 있다. 이 임상시험을 날려버릴 뻔한 자연재해가 있었다. 당시 임상시험 데이터를 사이트(site)에서 전화선으로, 휴렛팩커드(Hewlett-Packard, HP)에서 생산한 마이크로컴퓨터(micro-computer)에 직접 올리는 원격 데이터 입력 시스템(remote data entry system)이 처음 시작되었다. 필자는 페이퍼 데이터 관리(paper data management)를 주장했으나 필자의 의견보다는 컴퓨터 책임자들의 의견이 우세했다. 필자는 아직 검증되지 않은 IT 기술에 중대한 임상시험을 맡길 수 없다는 입장이었고 컴퓨터 전문가들은 HP를 못 믿으면 누구를 믿겠냐는 입장이었다. HP는 당시 세계 최고의 전자기기 회사였다. 결국 필자도 사이트에서 페이퍼로 데이터를 백업하는 조건으로 합의했다. 약 2년간 잘 진행되었고 필자도 할 말이 없었다. 필자의 관여에 대해 쓸데없는 짓이라는 힐난이 있었지만 데이터는 백업 되어야 한다는 입장을 고수했다. 1984년 여름으로 기억한다. 워싱턴 D.C.에 기록적 폭풍과 폭우가 닥쳤다. 천둥 번개가 밤새 계속되었고 정전사태가 벌어졌다. 다음 날 아침, 출근하자마자 확인해 보니 HP 마이크로 컴퓨터 디스크(micro computer disc)가 백업 디스크(back-up disc)와 더불어 모두 깨져 있었다. 그 동안 수집된 데이터가 모두 날아간 것이다. 밤새 친 천둥번개로 인한 파워 서지(power surge)가 원인이라고 컴퓨터 담당자가 설명했다. 연구소(Institute)에서는 난리가 났다. 필자는 페이퍼로 사이트에 백업이 되어 있으니 걱정하지 말라고 주변을 안심시키고, 페이퍼 데이터 관리를 다시 시작함으로써 임상시험을 성공리에 마칠 수 있었다. 필자는 이 임상시험을 성공리에 마쳐서 연구소(Institute)에서 표창을 받고 미국 국립 아동 건강 및 인간 발달 연구소(National Institute of Child Health and Human Development)에서 Biometry Chief를 제의 받고 자리를 옮겼다. 이번에는 4500명을 대상으로 하는 임신중독증 관련 임상시험이 기다리고 있었다. 임상시험이 자연재해에 취약함을 필자는 몸소 경험했다. 미국에서는 인터넷이 개발되면서 1990년대 중반부터 EDC(Electronic Data Capture)가 도입되었지만 2000년도 초기만 해도 페이퍼 백업(paper backup)을 요구하였다. 이제는 페이퍼 백업은 모두 사라졌다. 그러나 자연재해에 의한 임상시험의 취약점이 사라진 것은 아니다. 임상시험은 전 세계에서 진행되고 있다. 국내 바이오-제약사들의 많은 임상시험 역시 전 세계에서 진행되고 있다. 임상시험에 미칠 자연재해가 남의 일이 아니다. 자연재해 뿐 아니다. 인재(人災) 역시 심각한 문제다. 2018년 11월 KT 아현지사의 화재 사건, 2022년 10월 카카오 데이터 센터의 화재 사건 등은 전형적 인재다. 이런 인재도 임상시험을 파괴할 수 있다. 자연재해의 몇 가지 사례를 보자. 1990년 9월 수해로 아산병원이 침수되었다. 2001년 6월 미국 텍사스 대학교 건강과학센터(University of Texas Health Science Center)의 임상연구 시설과 IRB 사무실이 열대성 폭풍 앨리슨(Allison)으로 침수되었다. 2005년 뉴올리언즈 옥스너 의료센터(Ochsner Medical Center)에서 허리케인 카트리나(Katrina)로 인해 항암 임상시험 중인 입원 환자들이 미국내 여러 시설로 긴급 철수해야 했다. 2011년 뉴질랜드 크라이스트처치(Christchruch) 지진으로 병원이 파괴되고 연속되는 여진으로 몇 개월간 병원에 접근을 할 수 없었다. 푸에르토리코는 거의 상시로 발생하는 허리케인 위협으로 인해 임상시험 계획에 비상 대책을 포함한다. 우리나라도 아산병원 침수사건 이래 행정안전부는 지하공간 침수방지를 위한 수방기준 실무매뉴얼을 발행했다. 허리케인 카트리나를 겪은 루이지애나 주립대학교(Louisiana State University) 연구자들은 자연재해에 대비하여 두 가지를 강조했다. 첫째는 임상시험에 참여하는 환자와 커뮤니케이션 플랜을 준비할 것을 강조했다. 둘째는 사이트에 닥칠 재해에 대비해 리서치 데이터의 안전성 대책을 세울 것을 강조했다. 마이애미 대학교(University of Miami)에서는 허리케인대비 임상연구 매뉴얼을 발행했다. 연구자가 준비해야 할 사항들, 임상연구 참여자들을 위한 고려사항, 임상시험 중 수집되는 샘플 보호 등에 대한 비상 대책, 데이터 보호대책, 자연재해 후에 임상연구 연속성에 대한 대책 등으로 구성되어 있다. 미국 FDA는 2023년 9월 재난(Disasters)과 공중건강위기(Public Health Emergencies)로 인한 혼란상황에서의 임상시험 진행에 관한 지침(guidance)을 발행했다. FDA 지침의 목적은 위기와 혼란상황에서 임상참여자들을 보호하고, 임상시험 관리기준(GCP)를 유지하고, 임상시험에 미칠 위험을 최소화할 수 있도록 스폰서, IRB, 연구자들을 지원하는 데 있다고 했다. 그리고 Q&A에서 22개의 상황을 상정해 대응 방안에 대하여 논의하고 있다. 임상시험 데이터는 자연재해 또는 인재로부터 보호받기 위한 대책이 철저하게 준비되어야 한다. 2022년 태풍 '힌남노'로 포항제철은 135일간 생산공정이 중단되었고 그 손해가 2조가 넘었다고 한다. 우리는 해마다 태풍, 폭설 등 자연재해와 끊임없는 화재로 많은 피해를 입고 있다. 임상시험도 자연재해와 인재에 취약하다는 사실을 염두에 두고 대비책을 세우는 것이 중요할 것 같다. 더욱이 국내 제약 바이오사들도 MRCTs(Multi-Regional Clinical Trials, 다지역·다국가 임상시험)를 많이 진행하고 있다. 재해는 시간과 장소를 가리지 않는다. 각종 재해를 대비해야 할 것이다.2024-12-16 08:50:19데일리팜 -
 3세 경영 동성제약, 자금조달 예고…투자활동 강화[데일리팜=이석준 기자] 3세 경영에 돌입한 동성제약이 자금조달을 예고했다. 이를 통해 재무 구조 개선 및 투자 활동(R&D, 타법인 지분 취득 등)을 강화하려는 움직임으로 풀이된다. 동성제약은 26일 열린 임시주주총회에서 원용민씨를 사내이사로 신규 선임했다. 또 전환사채(CB)와 신주인수권부사채(BW) 발행 상한금액을 기존 400억원에서 2000억원으로 확대했다. 원용민씨는 EY 한영 회계법인 감사본부, 수앤파이낸셜인베스트먼트 PE본부에서 근무했고 현재는 밸류시스템자산운영 이사로 활동 중이다. 다년간 자금조달 및 투자운용 경험을 바탕으로 전문성을 보유한 재무전문가라는 평가를 받는다. 자금조달을 예고하는 대목이다. 재무전문가 영입과 사채 발행 상한금액을 확대했기 때문이다. 동성제약은 꾸준히 메자닌 자금을 조달하고 있다. 2016년 7월(납입일 기준) CB 100억원, 2018년 4월 CB 240억원, 2021년 8월 BW 85억원, 2023년 7월 BW 140억원 등이다. 이에 정관상 잔여 발행한도 상향이 필요했고 이번에 CB와 BW를 각 2000억원까지 확대하게 됐다. 2년마다 자금조달이 이뤄졌다는 점을 감안하면 조만간 자금 수혈이 이뤄질 것으로 보인다. 또 기존보다 자금조달 규모가 확대될 가능성도 점쳐진다. 실제 동성제약은 200억원 규모 CB 발행을 추진중으로 알려진다. 동성제약은 자금조달시 다방면에 활용할 수 있다. 먼저 재무 구조 개선이다. 올 9월말 현재 부채비율은 228.26%, 순차입금비율은 140.40%이다. 지난해말 부채비율(188.87%)과 순차입금비율(92.31%)보다 높아졌다. 보통 부채비율과 순차입금비율은 각각 100%, 20% 이하를 안정적으로 본다. 동성제약은 이를 상회하고 있다. 동성제약의 현금성자산은 7억원, 차입금(신주인수권부사채 포함)은 528억원이다. 차입금 대부분은 1년 이내 상환해야하는 단기차입금으로 유동성 관리가 필요한 상황이다. 연구개발이나 타법인 투자에 투입될 수도 있다. 동성제약은 올 4월 광역학 치료에 쓰이는 광과민제 '포노젠(DSP1944)'의 췌장암 2상을 승인받았다. 최근에는 포노젠 복막암 진단 IRB(임상윤리심의위원회)를 세브란스 병원에 신청했다. 모든 임상은 IRB 승인이 통과돼야 진행할 수 있다. 회사는 지난해 부동산 업체 디에스이엔에스 지분투자를 단행했다. 타법인 투자는 10여년 만으로 이를 기점으로 투자 활동을 강화할 수 있다. 업계 관계자는 "동성제약은 지난 10월부터 오너 3세 나원균 대표이사 부사장 체제다. 이번 임총서 재무전문가를 영입하고 자금조달 한도를 상향한 것도 나 대표의 의중이 담겨있다고 볼 수 있다"고 진단했다. 한편 나 대표는 1986년생으로 미국 에모리 대학교(Emory University)에서 응용수학과 및 경제학과를 복수전공하고, 이후 한국주택금융공사 및 금융위원회 등을 거쳐 2019년 동성제약에 입사했다. 이후 국제 전략실에서 해외 사업을 총괄하며 미주, 유럽, 동남아 등 글로벌 시장 매출을 2019년 42억에서 지난해 약 200억으로 성장시켰다. 2022년 사내이사 선임, 2024년 부사장으로 승진했다. 나 대표는 창업주 故 이선규 회장 외동딸 경희씨(동성제약 계열사 오마샤리프화장품 공동 대표, 송음의약재단 이사장, 이양구 전 대표 누나) 아들이자 이양구 전 대표의 조카다.& 160;2024-11-26 16:13:34이석준
3세 경영 동성제약, 자금조달 예고…투자활동 강화[데일리팜=이석준 기자] 3세 경영에 돌입한 동성제약이 자금조달을 예고했다. 이를 통해 재무 구조 개선 및 투자 활동(R&D, 타법인 지분 취득 등)을 강화하려는 움직임으로 풀이된다. 동성제약은 26일 열린 임시주주총회에서 원용민씨를 사내이사로 신규 선임했다. 또 전환사채(CB)와 신주인수권부사채(BW) 발행 상한금액을 기존 400억원에서 2000억원으로 확대했다. 원용민씨는 EY 한영 회계법인 감사본부, 수앤파이낸셜인베스트먼트 PE본부에서 근무했고 현재는 밸류시스템자산운영 이사로 활동 중이다. 다년간 자금조달 및 투자운용 경험을 바탕으로 전문성을 보유한 재무전문가라는 평가를 받는다. 자금조달을 예고하는 대목이다. 재무전문가 영입과 사채 발행 상한금액을 확대했기 때문이다. 동성제약은 꾸준히 메자닌 자금을 조달하고 있다. 2016년 7월(납입일 기준) CB 100억원, 2018년 4월 CB 240억원, 2021년 8월 BW 85억원, 2023년 7월 BW 140억원 등이다. 이에 정관상 잔여 발행한도 상향이 필요했고 이번에 CB와 BW를 각 2000억원까지 확대하게 됐다. 2년마다 자금조달이 이뤄졌다는 점을 감안하면 조만간 자금 수혈이 이뤄질 것으로 보인다. 또 기존보다 자금조달 규모가 확대될 가능성도 점쳐진다. 실제 동성제약은 200억원 규모 CB 발행을 추진중으로 알려진다. 동성제약은 자금조달시 다방면에 활용할 수 있다. 먼저 재무 구조 개선이다. 올 9월말 현재 부채비율은 228.26%, 순차입금비율은 140.40%이다. 지난해말 부채비율(188.87%)과 순차입금비율(92.31%)보다 높아졌다. 보통 부채비율과 순차입금비율은 각각 100%, 20% 이하를 안정적으로 본다. 동성제약은 이를 상회하고 있다. 동성제약의 현금성자산은 7억원, 차입금(신주인수권부사채 포함)은 528억원이다. 차입금 대부분은 1년 이내 상환해야하는 단기차입금으로 유동성 관리가 필요한 상황이다. 연구개발이나 타법인 투자에 투입될 수도 있다. 동성제약은 올 4월 광역학 치료에 쓰이는 광과민제 '포노젠(DSP1944)'의 췌장암 2상을 승인받았다. 최근에는 포노젠 복막암 진단 IRB(임상윤리심의위원회)를 세브란스 병원에 신청했다. 모든 임상은 IRB 승인이 통과돼야 진행할 수 있다. 회사는 지난해 부동산 업체 디에스이엔에스 지분투자를 단행했다. 타법인 투자는 10여년 만으로 이를 기점으로 투자 활동을 강화할 수 있다. 업계 관계자는 "동성제약은 지난 10월부터 오너 3세 나원균 대표이사 부사장 체제다. 이번 임총서 재무전문가를 영입하고 자금조달 한도를 상향한 것도 나 대표의 의중이 담겨있다고 볼 수 있다"고 진단했다. 한편 나 대표는 1986년생으로 미국 에모리 대학교(Emory University)에서 응용수학과 및 경제학과를 복수전공하고, 이후 한국주택금융공사 및 금융위원회 등을 거쳐 2019년 동성제약에 입사했다. 이후 국제 전략실에서 해외 사업을 총괄하며 미주, 유럽, 동남아 등 글로벌 시장 매출을 2019년 42억에서 지난해 약 200억으로 성장시켰다. 2022년 사내이사 선임, 2024년 부사장으로 승진했다. 나 대표는 창업주 故 이선규 회장 외동딸 경희씨(동성제약 계열사 오마샤리프화장품 공동 대표, 송음의약재단 이사장, 이양구 전 대표 누나) 아들이자 이양구 전 대표의 조카다.& 160;2024-11-26 16:13:34이석준 -
 동성제약, 암 진단 시장 본격 진입 시동[데일리팜=노병철 기자] 동성제약(대표 나원균)은 자체 개발한 광민감제 DSP-1944(포노젠)의 복막암 진단 IRB를 세브란스 병원에 신청했다고 18일 밝혔다. 동성제약이 신청한 IRB는 광민감제 포노젠을 암 진단용으로 추가 개발해 '복강 내 위암의 복막 전이 진단을 위한 광역학 진단의 효과 및 안정성 평가'를 위한 임상 시험이다. IRB는 임상 시험에 참여하는 대상자의 권리, 안전, 복지를 위해 시험 기관에 독립적으로 설치된 의결 기구로 모든 임상 시험은 IRB 승인이 통과돼야 진행할 수 있다. 이번 IRB 신청은 지난 10월, 신규 선임된 나원균 대표이사의 체제하에 진행된 첫 번째 PDD(광역학 진단) 관련 성과로, 회사의 PDT(광역학) 치료와 PDD 사업에 대한 강력한 의지를 나타낸다. 난치암인 복막암은 수술 전 CT 스캔과 기존 복강경 검사에서 놓치는 경우가 많아 진단에 어려움이 있는 질병으로 동성제약은 이미 토끼를 이용한 전임상 시험에서 광과민제 포노젠의 효과를 확인한 바 있다. 포노젠을 405nm에서 활성화한 광역학 진단(PDD)을 사용해 복강경 검사의 병기 진단 정확도를 평가한 결과, 진단율이 상승한 것을 확인했고 이를 근거로 복막암 환자 대상 임상시험에 본격적으로 진입할 예정이다. 동성제약 관계자는 "국내 유일 광역학 치료 및 진단 선도주자인 동성제약은 이번 IRB 신청을 통해 광역학 치료(PDT) 뿐만 아니라 진단(PDD) 부문에서도 글로벌 기업과 어깨를 나란히 할 수 있는 입지를 마련했다"고 전했다. 한편, 동성제약 포노젠의 복막암 진단 효과는 지난 국제복막암학회(PSOGI)는 물론, 올해 4월 세계 최대 암 학회인 미국암연구학회(AACR)에서 발표되어 그 우수성을 증명했다.2024-11-18 09:35:41노병철
동성제약, 암 진단 시장 본격 진입 시동[데일리팜=노병철 기자] 동성제약(대표 나원균)은 자체 개발한 광민감제 DSP-1944(포노젠)의 복막암 진단 IRB를 세브란스 병원에 신청했다고 18일 밝혔다. 동성제약이 신청한 IRB는 광민감제 포노젠을 암 진단용으로 추가 개발해 '복강 내 위암의 복막 전이 진단을 위한 광역학 진단의 효과 및 안정성 평가'를 위한 임상 시험이다. IRB는 임상 시험에 참여하는 대상자의 권리, 안전, 복지를 위해 시험 기관에 독립적으로 설치된 의결 기구로 모든 임상 시험은 IRB 승인이 통과돼야 진행할 수 있다. 이번 IRB 신청은 지난 10월, 신규 선임된 나원균 대표이사의 체제하에 진행된 첫 번째 PDD(광역학 진단) 관련 성과로, 회사의 PDT(광역학) 치료와 PDD 사업에 대한 강력한 의지를 나타낸다. 난치암인 복막암은 수술 전 CT 스캔과 기존 복강경 검사에서 놓치는 경우가 많아 진단에 어려움이 있는 질병으로 동성제약은 이미 토끼를 이용한 전임상 시험에서 광과민제 포노젠의 효과를 확인한 바 있다. 포노젠을 405nm에서 활성화한 광역학 진단(PDD)을 사용해 복강경 검사의 병기 진단 정확도를 평가한 결과, 진단율이 상승한 것을 확인했고 이를 근거로 복막암 환자 대상 임상시험에 본격적으로 진입할 예정이다. 동성제약 관계자는 "국내 유일 광역학 치료 및 진단 선도주자인 동성제약은 이번 IRB 신청을 통해 광역학 치료(PDT) 뿐만 아니라 진단(PDD) 부문에서도 글로벌 기업과 어깨를 나란히 할 수 있는 입지를 마련했다"고 전했다. 한편, 동성제약 포노젠의 복막암 진단 효과는 지난 국제복막암학회(PSOGI)는 물론, 올해 4월 세계 최대 암 학회인 미국암연구학회(AACR)에서 발표되어 그 우수성을 증명했다.2024-11-18 09:35:41노병철 -
 압타머사이언스 "연내 압타머 신약 임상 돌입"[데일리팜=이석준 기자] 압타머사이언스(대표이사 한동일)는 약 200억 원 규모의 주주배정 유상증자 증권신고서 효력이 발생했다고 밝혔다. 주당발행가액은 1617원이며 총 발행주식수는 123만 주로 납입일은 다음달 12일이다. 자금조달이 성공적으로 마무리될 경우 법인세비용 차감 전 계속사업 손실 비율이 50% 미만으로 개선돼 관리종목 지정 리스크가 해소될 전망이다. 압타머사이언스는 이번 유증을 통해 조달한 자금으로 기업의 재무구조 개선과 압타머 기반의 혁신 신약 파이프라인의 사업화에 집중할 예정이다. 회사 주력 제품인 ApDC® (Aptamer-Drug Conjugate, 압타머-약물 접합체) 간암 치료제 ‘AST-201’ 국내 임상을 연내 착수한다. 지난 10월 식품의약품안전처로부터 임상시험계획서(IND)를 승인 받은 이후 2개 병원으로부터 생명윤리심의위원회(IRB)를 통과했다. 사전에 각 병원의 시험책임자들과 일정을 조율해둔 덕분에 행정절차와 환자 모집까지 패스트트랙(fasttrack)으로 진행하고, 연말부터는 본격적으로 임상 투약을 시작한다는 계획이다.2024-11-08 09:50:19이석준
압타머사이언스 "연내 압타머 신약 임상 돌입"[데일리팜=이석준 기자] 압타머사이언스(대표이사 한동일)는 약 200억 원 규모의 주주배정 유상증자 증권신고서 효력이 발생했다고 밝혔다. 주당발행가액은 1617원이며 총 발행주식수는 123만 주로 납입일은 다음달 12일이다. 자금조달이 성공적으로 마무리될 경우 법인세비용 차감 전 계속사업 손실 비율이 50% 미만으로 개선돼 관리종목 지정 리스크가 해소될 전망이다. 압타머사이언스는 이번 유증을 통해 조달한 자금으로 기업의 재무구조 개선과 압타머 기반의 혁신 신약 파이프라인의 사업화에 집중할 예정이다. 회사 주력 제품인 ApDC® (Aptamer-Drug Conjugate, 압타머-약물 접합체) 간암 치료제 ‘AST-201’ 국내 임상을 연내 착수한다. 지난 10월 식품의약품안전처로부터 임상시험계획서(IND)를 승인 받은 이후 2개 병원으로부터 생명윤리심의위원회(IRB)를 통과했다. 사전에 각 병원의 시험책임자들과 일정을 조율해둔 덕분에 행정절차와 환자 모집까지 패스트트랙(fasttrack)으로 진행하고, 연말부터는 본격적으로 임상 투약을 시작한다는 계획이다.2024-11-08 09:50:19이석준 -
 드림CIS, 의대교수 초청 질환별 임상시험 최신 지견 공유[데일리팜=이탁순 기자] 드림씨아이에스(대표 유정희)가 '드림사이언스 아카데미(DreamScience Academy)' 프로그램 진행을 통해 임상시험 시장 인재 육성 및 임상 품질 제고에 나섰다. 드림사이언스 아카데미(이하 드림아카데미)는 각 질환별 다양한 진료교수로부터 신약 개발 및 임상시험에 대한 최신 지견과 관련된 강연을 듣는 자리로, CRO업계에서는 최초로 도입돼 운영하고 있다. 드림아카데미 프로그램을 통해 사내 임직원들은 물론 국내 제약회사 및 바이오벤처의 임상 및 개발본부 관계자를 초청해 해당 질환에 대한 전문지식을 높이고 최신의 업계 트렌드를 소개, 신약개발 및 임상시험 진행에 도움을 주는 것을 목적으로 하고 있다. 지난 14일, 고려대학교 안암병원의 이수현 종양내과 교수가 드림씨아이에스 본사를 방문해 '정밀 의료 시대의 임상시험 디자인'이라는 주제로 특강을 진행했다. 이수현 교수는 의뢰사 및 연구자 입장을 모두 겪어본 경험을 바탕으로 정밀 임상 디자인 시 문제점을 다루고, 해외 모델을 벤치마킹한 사례를 소개하며 강연을 진행했다. 특히 글로벌 제약사에서 근무했던 경험 및 연구자로서 진행한 임상 사례 등을 소개하며 임상시험 디자인 시 꼭 필요한 점들을 강조했다. 드림아카데미는 올해 1월부터 배균섭 교수(서울아산병원), 윤영철 교수(대한치매학회장), 정근화 교수(서울대 신경과), 정영훈 교수(순환기의공학회 이사장), 송정수 교수(대한류마티스학회장), 유승영 교수(경희대 안과), 임수 교수(분당서울대학교병원) 등 각기 다른 분야에서 주요 교수들이 참여했다. 앞으로 IRB 실례, 알츠하이머 병, 뇌졸중, 망막질환, 류마티스질환, 뇌질환, 비만 및 내분비질환 등 다양한 질환에 대한 임상현황에 대해 알아본 바 있으며, 추후 ▲DERM(탈모·미용) ▲FM-Obesity ▲세포치료 ▲첨단혁신 치료 신약 등의 주제가 다뤄질 예정이다. 드림씨아이에스의 한 관계자는 "아카데미 프로그램을 통해 당사 임상 실무진은 물론 의뢰사로부터 긍정적인 피드백을 지속적으로 받고 있다. 기존에는 임상시험을 실제로 진행하는 교수님(연구자)의 입장과 실례 등을 쉽게 들을 수 없었지만, 본 프로그램을 통해 접근성이 용이해 졌다는 의견과 함께 임상 진행 시 다양한 참고 의견으로도 활용 가능해 향후 임상 퀄리티 향상에도 긍정적인 영향을 미칠 것으로 예상된다"고 전했다.2024-10-25 14:42:46이탁순
드림CIS, 의대교수 초청 질환별 임상시험 최신 지견 공유[데일리팜=이탁순 기자] 드림씨아이에스(대표 유정희)가 '드림사이언스 아카데미(DreamScience Academy)' 프로그램 진행을 통해 임상시험 시장 인재 육성 및 임상 품질 제고에 나섰다. 드림사이언스 아카데미(이하 드림아카데미)는 각 질환별 다양한 진료교수로부터 신약 개발 및 임상시험에 대한 최신 지견과 관련된 강연을 듣는 자리로, CRO업계에서는 최초로 도입돼 운영하고 있다. 드림아카데미 프로그램을 통해 사내 임직원들은 물론 국내 제약회사 및 바이오벤처의 임상 및 개발본부 관계자를 초청해 해당 질환에 대한 전문지식을 높이고 최신의 업계 트렌드를 소개, 신약개발 및 임상시험 진행에 도움을 주는 것을 목적으로 하고 있다. 지난 14일, 고려대학교 안암병원의 이수현 종양내과 교수가 드림씨아이에스 본사를 방문해 '정밀 의료 시대의 임상시험 디자인'이라는 주제로 특강을 진행했다. 이수현 교수는 의뢰사 및 연구자 입장을 모두 겪어본 경험을 바탕으로 정밀 임상 디자인 시 문제점을 다루고, 해외 모델을 벤치마킹한 사례를 소개하며 강연을 진행했다. 특히 글로벌 제약사에서 근무했던 경험 및 연구자로서 진행한 임상 사례 등을 소개하며 임상시험 디자인 시 꼭 필요한 점들을 강조했다. 드림아카데미는 올해 1월부터 배균섭 교수(서울아산병원), 윤영철 교수(대한치매학회장), 정근화 교수(서울대 신경과), 정영훈 교수(순환기의공학회 이사장), 송정수 교수(대한류마티스학회장), 유승영 교수(경희대 안과), 임수 교수(분당서울대학교병원) 등 각기 다른 분야에서 주요 교수들이 참여했다. 앞으로 IRB 실례, 알츠하이머 병, 뇌졸중, 망막질환, 류마티스질환, 뇌질환, 비만 및 내분비질환 등 다양한 질환에 대한 임상현황에 대해 알아본 바 있으며, 추후 ▲DERM(탈모·미용) ▲FM-Obesity ▲세포치료 ▲첨단혁신 치료 신약 등의 주제가 다뤄질 예정이다. 드림씨아이에스의 한 관계자는 "아카데미 프로그램을 통해 당사 임상 실무진은 물론 의뢰사로부터 긍정적인 피드백을 지속적으로 받고 있다. 기존에는 임상시험을 실제로 진행하는 교수님(연구자)의 입장과 실례 등을 쉽게 들을 수 없었지만, 본 프로그램을 통해 접근성이 용이해 졌다는 의견과 함께 임상 진행 시 다양한 참고 의견으로도 활용 가능해 향후 임상 퀄리티 향상에도 긍정적인 영향을 미칠 것으로 예상된다"고 전했다.2024-10-25 14:42:46이탁순 -
 뉴로바이오젠, 알츠하이머 신약 후보 2상 임상 진입[데일리팜=이혜경 기자] 국내 업체가 개발 중인 알츠하이머 신약이 2상 임상시험에 들어간다. 식품의약품안전처는 30일 뉴로바이오젠이 신청한 '경도인지장애 단계의 알츠하이머병 및 경도 알츠하이머병 치매 환자를 대상으로 KDS2010의 유효성 및 안전성을 평가하기 위한 무작위배정, 이중 눈가림, 위약 대조, 용량탐색, 제2a상 임상시험'을 승인했다. 이번 임상시험은 건국대병원에서 진행된다. 신약 후보물질 KDS2010는 먹는(경구용) 약으로 개발 중이며, 기존 피하주사 제형보다 복약 투약성이 뛰어나고 오심 구토 등 소화기계 부작용을 줄일 것으로 기대되는 약물이다. KDS2010는 오랫동안 파킨슨 치료 타깃으로 여겨지고 있는 마오비(MAO-B, 모노아민 산화효소-B) 효소를 표적하는 새로운 기전의 치료제다. 퇴행성 뇌질환치료제로써 국제학술지 Science Advances 연구결과를 보면 KDS2010는 기존 약물이 장기간 투여 시 약물에 의해 완전히 저해된 마오비 효소의 역할을 대신할 수 있는 다오(DAO, diamine oxidase) 효소가 과발현 되면서 다시 '가바(GABA)'가 과생성 되고, 이러한 생체 내 대체기전으로 인해 인지기능 개선 효능이 사라진다는 것이 검증됐다. 이 논문을 토대로 뉴로바이오젠의 후보약물은 장기간 투약해도 이러한 대체기전을 작동하지 않아 인지기능이 현저히 개선된다는 결과를 확인했다고 밝혔다. 여기에 KDS2010은 GABA 과생성에 기인한 기전으로 알츠하이머병 외에도 뇌졸중, 척수손상, 비만치료제로의 임상적 적응증 확대가 가능하다는 특징을 가지고 있다. 이러한 학술적 특징을 전제로 원숭이를 포함한 비임상 독성 효력 시험을 진행한 결과, KDS2010은 뇌질환(치매, 알츠하이머) 약물의 필수허가요건인 생체 독성 및 다른 신경계에 부작용이 없는 안전성을 보였다. 뉴로바이오젠은 지난해 12월 11일자로 서울대병원에서 진행한 KDS2010의 안전성 및 내약성 1상 IRB 종료 보고를 완료했다. 1상은 건강한 성인 남녀는 물론 코카시안 대상자 50% 포함해 미국 FDA 및 글로벌 임상 규모와 수준에 부합한 규모로 진행했다. 이번 2상은 1상 데이터를 바탕으로 수행된다. 한편 바이오 및 의료기기 전문기업 시너지이노베이션은 2019년 11월 신약개발 파이프라인을 구축하기 위해 뉴로바이오젠 전환사채(CB)에 투자했다. CB 전환 시 최대주주의 지위를 확보하게 된다.2024-08-30 16:48:50이혜경
뉴로바이오젠, 알츠하이머 신약 후보 2상 임상 진입[데일리팜=이혜경 기자] 국내 업체가 개발 중인 알츠하이머 신약이 2상 임상시험에 들어간다. 식품의약품안전처는 30일 뉴로바이오젠이 신청한 '경도인지장애 단계의 알츠하이머병 및 경도 알츠하이머병 치매 환자를 대상으로 KDS2010의 유효성 및 안전성을 평가하기 위한 무작위배정, 이중 눈가림, 위약 대조, 용량탐색, 제2a상 임상시험'을 승인했다. 이번 임상시험은 건국대병원에서 진행된다. 신약 후보물질 KDS2010는 먹는(경구용) 약으로 개발 중이며, 기존 피하주사 제형보다 복약 투약성이 뛰어나고 오심 구토 등 소화기계 부작용을 줄일 것으로 기대되는 약물이다. KDS2010는 오랫동안 파킨슨 치료 타깃으로 여겨지고 있는 마오비(MAO-B, 모노아민 산화효소-B) 효소를 표적하는 새로운 기전의 치료제다. 퇴행성 뇌질환치료제로써 국제학술지 Science Advances 연구결과를 보면 KDS2010는 기존 약물이 장기간 투여 시 약물에 의해 완전히 저해된 마오비 효소의 역할을 대신할 수 있는 다오(DAO, diamine oxidase) 효소가 과발현 되면서 다시 '가바(GABA)'가 과생성 되고, 이러한 생체 내 대체기전으로 인해 인지기능 개선 효능이 사라진다는 것이 검증됐다. 이 논문을 토대로 뉴로바이오젠의 후보약물은 장기간 투약해도 이러한 대체기전을 작동하지 않아 인지기능이 현저히 개선된다는 결과를 확인했다고 밝혔다. 여기에 KDS2010은 GABA 과생성에 기인한 기전으로 알츠하이머병 외에도 뇌졸중, 척수손상, 비만치료제로의 임상적 적응증 확대가 가능하다는 특징을 가지고 있다. 이러한 학술적 특징을 전제로 원숭이를 포함한 비임상 독성 효력 시험을 진행한 결과, KDS2010은 뇌질환(치매, 알츠하이머) 약물의 필수허가요건인 생체 독성 및 다른 신경계에 부작용이 없는 안전성을 보였다. 뉴로바이오젠은 지난해 12월 11일자로 서울대병원에서 진행한 KDS2010의 안전성 및 내약성 1상 IRB 종료 보고를 완료했다. 1상은 건강한 성인 남녀는 물론 코카시안 대상자 50% 포함해 미국 FDA 및 글로벌 임상 규모와 수준에 부합한 규모로 진행했다. 이번 2상은 1상 데이터를 바탕으로 수행된다. 한편 바이오 및 의료기기 전문기업 시너지이노베이션은 2019년 11월 신약개발 파이프라인을 구축하기 위해 뉴로바이오젠 전환사채(CB)에 투자했다. CB 전환 시 최대주주의 지위를 확보하게 된다.2024-08-30 16:48:50이혜경
-
 약제부 야간전담, 주말(토)전담 약사 모집
약제부 야간전담, 주말(토)전담 약사 모집 -
 [부산 메리놀병원] 정규약사 및 주말 오전파트 약사 모집
[부산 메리놀병원] 정규약사 및 주말 오전파트 약사 모집 -
 제제연구 기술영업 부장급 채용(경력 10년↑)
제제연구 기술영업 부장급 채용(경력 10년↑) -
 2026년도 약사(야간약사, 주말약사, 기간제약사) 모집 공고
2026년도 약사(야간약사, 주말약사, 기간제약사) 모집 공고 -
 국립암센터 전공약사 채용
국립암센터 전공약사 채용 -
 경남 함안공장 품질관리약사
경남 함안공장 품질관리약사 -
 근무약사님 모십니다
근무약사님 모십니다 -
 퍼스트병원에서 약사님 모십니다
퍼스트병원에서 약사님 모십니다 -
 BD라이센싱팀 관리자/팀장, 신입/경력사원 및 전문연구요원 모집
BD라이센싱팀 관리자/팀장, 신입/경력사원 및 전문연구요원 모집 -
 ★ 약제과 오후 파트 정규직 약사 초빙 ★
★ 약제과 오후 파트 정규직 약사 초빙 ★ -
 에이스병원 상근(주40시간) 약사님 모십니다.
에이스병원 상근(주40시간) 약사님 모십니다. -
 '정규직 약사' '야간약사' '주말약사' 채용
'정규직 약사' '야간약사' '주말약사' 채용 -
 [중앙보훈병원] 약제실 약사 채용
[중앙보훈병원] 약제실 약사 채용 -
 2026년도 제1회 식품의약품안전처 공무원 경력경쟁채용
2026년도 제1회 식품의약품안전처 공무원 경력경쟁채용 -
 정신건강의학과 병원 약제과장님을 초빙합니다.
정신건강의학과 병원 약제과장님을 초빙합니다. -
 삼성서울병원 (주말전담) 약제부 계약직 약사 채용
삼성서울병원 (주말전담) 약제부 계약직 약사 채용 -
 [한독] 신입 및 경력 직무별 수시채용 (마감일 직무별 상이)
[한독] 신입 및 경력 직무별 수시채용 (마감일 직무별 상이) -
 [한국오츠카제약] CTM(계약직) 채용
[한국오츠카제약] CTM(계약직) 채용 -
 [경기도 광주] 복산나이스 관리약사 채용
[경기도 광주] 복산나이스 관리약사 채용 -
 RA, MSL , Head of Market Access, Head of Medical & Regulatory 등 각 부문 모집
RA, MSL , Head of Market Access, Head of Medical & Regulatory 등 각 부문 모집 -
 약제부 정규직(신입) 약사 모집 공고
약제부 정규직(신입) 약사 모집 공고 -
 현대병원 평일정규 약사
현대병원 평일정규 약사 -
 아산충무병원 약제과 정규직 약사 모집
아산충무병원 약제과 정규직 약사 모집 -
 백암공장 QA약사 채용 경력무관
백암공장 QA약사 채용 경력무관 -
 향남&안성 제조관리 약사 채용(신입우대)
향남&안성 제조관리 약사 채용(신입우대) -
 Regulatory Affairs Specialist ( 16-month contract)
Regulatory Affairs Specialist ( 16-month contract) -
 로뎀요양병원 약사님 모십니다.
로뎀요양병원 약사님 모십니다. -
 [충남대학교병원] 정규직 약사 공개채용
[충남대학교병원] 정규직 약사 공개채용 -
 약제부 약사 채용 (평일/주말)
약제부 약사 채용 (평일/주말)

약국e몰

![[신신] 새사래 상처연고 습윤밴드](https://cdn.platpharm.co.kr/2025/10/2510210339570001784.webp)
![[더본메디칼] ATC인쇄리본 특가](https://cdn.platpharm.co.kr/2025/04/2504100527360001454.jpg)
![[셀로맥스] 베베락스 온가족 안심 관장약](https://cdn.platpharm.co.kr/2025/09/2509171131320018843.webp)
![[유한양행] 콘택콜드 걸렸구나 생각되면](https://cdn.platpharm.co.kr/2025/10/2510282252420008436.webp)
![[유한양행] 미녹펜겔 탈모스팟 집중케어](https://cdn.platpharm.co.kr/2025/09/2509220824180004563.webp)
![[SK케미칼] 속편한정 복합소화제](https://cdn.platpharm.co.kr/2025/12/2512040916400005920.webp)
![[리쥬올]레티노 멜라세럼 저자극 레티놀](https://cdn.platpharm.co.kr/2025/09/2509260219360000145.webp)
![[SK케미칼] 트라스트패취 피록시캄 성분](https://cdn.platpharm.co.kr/2025/10/2510020656150002375.webp)
![[신신] 아렉스 두번효과로 강력한](https://cdn.platpharm.co.kr/2025/10/2510230254510000664.webp)
![[리쥬올]리쥬올 PDRN 약국 1위 PDRN](https://cdn.platpharm.co.kr/2025/09/2509260220180000170.webp)
![[종근당] 벤포벨에스 어른들의 피로회복제](https://cdn.platpharm.co.kr/2025/07/2507290841210004645.webp)
![[아워팜] 건강한 힘, 바로바이오틱스 kids 비피더스 50억](https://i.baropharm.com/products/202511/1763106585375.png)
![[아워팜] 혈행건강 히어로, 혈행건강 초임계 rTG 오메가3 프리미엄](https://i.baropharm.com/products/f724f191-7052-4ad5-bc92-cda889cf13a6.png)
![[아워팜] 우리아이 맞춤설계, 바로타민 kids 엘더베리맛](https://i.baropharm.com/products/8eae7689-61be-4b71-9608-05364b05ea12.png)
![[한독] 붙이는 통증 전문가, 케토톱 액티브 플라스타(쿨) 40매](https://i.baropharm.com/products/202503/1741829602305.png)
![[레킷코리아] 목 아플 때, 스트렙실 허니&레몬 트로키 12정](https://i.baropharm.com/products/202502/1739520767049.png?label=PLAN_01)
![[휴온스 ] 비듬을 한번에, 니조랄 2%액](https://i.baropharm.com/products/478a284d-4361-4b4a-8a00-8bab80f34319.png?label=PLAN_01)
![[켄뷰] 다양한 통증에, 타이레놀정 500mg 10정](https://i.baropharm.com/products/6c6ea4f4-7ab2-44f2-a165-f062d80f525b.png)
![[오펠라] 부드럽고 편안한, 둘코락스에스장용정 20정](https://i.baropharm.com/products/202511/1762260404625.png)
![[켄뷰] 오리지널 폼타입, 로게인5%폼에어로졸60g](https://i.baropharm.com/products/dc84d96e-d0b4-46bc-bcc8-d62016406fe4.png)
![[아워팜] CJ웰케어, 바이오코어 1000억 유산균](https://i.baropharm.com/products/202512/1765955416559.png)
![[아워팜] 에너지 바로 충전, 바로콤](https://i.baropharm.com/products/202512/1764922282624.png)
![[아워팜] 아이들이 먼저찾는, 바로타민 kids 미네랄](https://i.baropharm.com/products/202512/1766121243228.png)

오늘의 TOP 10
- 1HK이노엔 미 파트너사, '케이캡' FDA 허가 신청
- 2[기자의 눈] JPM, K-제약 '참가의 시대' 끝났다
- 3피타바스타틴 허가 역대 최다...분기 1천억 시장의 매력
- 4성장은 체력 싸움…제약사 경쟁, 신뢰로 갈린다
- 5SK바사·롯바도 입성…송도, 바이오 '시총 156조' 허브로
- 6메가팩토리약국, 프랜차이즈 사업 진출…전국 체인화 시동
- 7'혼합음료 알부민' 1병당 단백질 1g뿐…"무늬만 알부민"
- 8동구바이오제약, 박종현 부사장 영입…미래전략부문 강화
- 9예상청구액 2300억 키트루다 급여 확대...건보재정 경고등
- 10권영희 회장 "한약사 문제 해결·성분명 법제화 올해 완수"
-
순위상품명횟수
-
1타이레놀정500mg(10정)30,426
-
2판콜에스내복액16,732
-
3텐텐츄정(10정)13,671
-
4까스활명수큐액12,867
-
5판피린큐액12,851