- LOGIN
- MemberShip
- 2026-05-03 21:03:14

- Policy
- Approval for PH-ILD inhalation drug 'Tyvaso' expected soon
- by Lee, Hye-Kyung Jun 11, 2024 05:48am
- Tyvaso Inhalation Solution. The approval of 'Tyvaso Inhalation Solution 0.6 mg/mL (treprostinil)' in South Korea is expected soon. It is approved in the United States as the treatment for pulmonary arterial hypertension (PAH) and pulmonary hypertension associated with interstitial lung disease (PH-ILD). According to industry sources on the 11th, the safety and effectiveness evaluation of Anterogen for the approval had been completed. The completion of the safety and efficacy evaluation is followed by the NDA. United Therapeutics’s Tyvaso is a treatment for patients with PH-ILD, and Anterogen has a domestic license. In August last year, the Ministry of Food and Drug Safety (MFDS) designated the drug as the 14th Global Innovative products on Fast Track (GIFT). Following the designation, Tyvaso has been under expedited review for approval. For GIFT-designated products, the review duration is shortened by 25% (i.e., 120→90 working days), rolling review is applied, which enables review on prepared and available documents, close communication between the reviewer and the developing company, such as product briefing and explanation of supplementation, is offered, and various regulatory supports, including RA consulting for fast commercialization, are provided. The company has applied for the efficacy and effectiveness of Tyvaso in PH-ILD, which is a fatal disease with no available treatments despite a survival rate of about 30% within three years of diagnosis. Tyvaso first received approval from the U.S. FDA in 2009 and received expanded indications for the first treatment of PH-ILD in 2021. The approval was based on the INCREASE clinical trial data. The trial was the largest, enrolling 326 patients with PH-ILD and the most comprehensive. Tyvaso met the primary endpoint and significantly improved 6-minute walk distance (6MWD) results. The treatment showed benefits in various major subgroups related to etiology, disease severity, age, sex, blood kinetics, and doses. Improvements in the secondary endpoints included the reduction of NT-proBNP levels, a heart biomarker, the time to clinical worsening, the change in the highest 6MWD at week 12, and the change in the lowest 6MWD at week 15.
- Company
- GC's Sanfilippo syndrome drug receives fast-track status
- by Son, Hyung-Min Jun 11, 2024 05:47am
- GC Biopharma announced on Tuesday that the U.S. FDA has granted Fast Track Designation for GC1130A, a treatment for Sanfilippo syndrome type A (MPS IIIA) that it has been co-developing with Novel Pharma. The fast-track designation follows the FDA's clearance of the Phase I investigational new drug (IND) application for GC1130A last month and is expected to further accelerate the development of GC1130A. Sanfilippo syndrome (type A) is a rare genetic disorder that causes central nervous system damage through the accumulation of heparan sulfate, leading to progressive neurological decline. Without treatment, patients face life-threatening complications by the age of 15. GC1130A is a new biological drug that is being developed using GC Biopharma’s high-concentration protein formulation technology, designed for administration to the central nervous system. It is delivered directly into the brain's ventricles through intracerebroventricular (ICV) injection, a method first applied globally by GC Biopharma's Hunter syndrome treatment, 'Hunterase', which has received marketing authorization in Japan. The potential of GC1130A to meet the unmet medical needs of Sanfilippo syndrome has been recognized by major drug regulatory agencies; in 2023, the FDA granted GC1130A Rare Pediatric Disease Designation (RPDD) and Orphan Drug Disease (ODD), and earlier this year, the European Medicines Agency (EMA) also granted GC1130A ODD status. Currently, GC Biopharma and Novel Pharma are preparing to initiate a multinational clinical trial to evaluate the safety and tolerability of GC1130A in Korea, the US, and Japan. A GC Biopharma official said, “With no approved treatment available for Sanfilippo syndrome, we are pleased that the FDA has granted GC1130A Fast Track designation. This designation will allow us to accelerate the development of our drug to bring hope to patients suffering from Sanfilippo syndrome.” The FDA's Fast Track program is designed to expedite the development and review of drugs intended to treat serious or unmet medical needs. The Fast Track designation provides extensive support, including frequent meetings with the FDA throughout the drug development, clinical, and approval stages.
- Policy
- Handok paces its efforts to reimburse Pivlaz in KOR
- by Lee, Tak-Sun Jun 11, 2024 05:47am
- Handok is taking a break from its pursuit of reimbursement for ‘Pivlaz,’ a new drug used to prevent cerebral vasospasm in patients with subarachnoid hemorrhage that the company is supplying and distributing in Korea. The company immediately applied for Pivlaz’s reimbursement to the Health Insurance Review and Assessment Service after receiving approval in December last year, but was found to have voluntarily withdrawn the application recently. The company is expected to realign its strategy and then pursue reimbursement again. According to industry sources on the 10th, Handok submitted a letter requesting the withdrawal of the application for the drug determination (reimbursement) of Pivlaz Inj (clazosentan). The drug was granted marketing authorization last year on December 7. It is a selective endothelin A receptor antagonist indicated for the prevention of cerebral vasospasm, vasospasm-related cerebral infarction, and cerebral ischemic events in adults who have undergone clipping surgery or coiling treatment for aneurysmal subarachnoid hemorrhage. Pivlaz is the first drug approved in Korea to prevent both cerebral vasospasm and its complications. Cerebral vasospasm, which occurs after aneurysmal subarachnoid hemorrhage, has been shown to double the risk of death in patients. It is also accompanied by serious complications such as localized paralysis, speech impairment, and decreased consciousness. Active prevention and treatment are crucial, but the field has been experiencing difficulties due to the lack of appropriate drugs. This is why Pivlaz is rising as a promising treatment option. Nxera Pharma Korea owns the sales rights for Pivlaz in Korea. The Japanese multinational company, former Sosei Heptares, changed its name to Nxera in April last year after acquiring the Swiss pharmaceutical company Idorsia, which developed Pivlaz. On April 12, Handok announced that it signed an exclusive domestic supply and distribution agreement with Nxera Pharma Korea for Pivlaz. Handok has been collaborating with Nxera Pharma Korea Korea since 2008 to conduct domestic clinical trials and obtain marketing authorization for Pivlaz. Handok and Nxera Pharma Korea are seeking to launch Pivlaz early next year. There is still time to receive reimbursement within the timeline. The industry’s attention is focused on whether the two companies will be able to realign their strategy and speed up the reimbursement process for Pivlaz.
- Company
- 'Novel drugs·CDMO competitiveness↑'
- by Son, Hyung-Min Jun 10, 2024 05:41am
- Major biotech companies in Korea participated in the BIO International Convention (BIO USA 2024) and introduced their in-house competitiveness. Over 50 Korea-based companies attended the BIO USA 2024, held between June 3-6 in San Diego, United States. They sought opportunities to expand partnerships along with discussions for technology transport. Samsung Biologics showcased its new platform, showing a strong directive to expand the company’s growth related to intensified cell-culture with improved manufacturing capacity. Lotte Biologics, ST Pharm, and Prestige Biologic introduced their contract development and manufacturing organization (CDMO) capacities and held partnering meetings for contract orders. Korean companies have made significant achievements in novel drug development. VaxCell Biotherapeutics showcased positive results of its novel candidate under a phase 2 clinical trial for hepatocellular carcinoma. Genome & Company won a technology transport contract for its antibody-drug conjugate (ADC), confirming its competitiveness. China absent from the BIO USA…all eyes on K-bio According to industry sources on June 9th, 47 Korean companies ran booths during the BIO USA, which was 6 more companies than last year. BIO USA is the world’s largest biopharmaceutical convention, with more than 20,000 leaders in the biopharmaceutical industry participating. Continuing from last year, Korean CDMO companies have gathered attention this year. Notably, due to the introduction of the Biosecure Act by the United States, all eyes were drawn to the Korean companies in the absence of WuXi Biologics, China’s largest CDMO. The United States aims to limit the business with the Chinese biotech companies. Therefore, the analysis suggests that along with Swiss-based Lonza and Japan-based Fujifilm, Korean CDMO companies will have more opportunities. The Korea booth at the BIO International Convention (BIO USA 2024) (photo=KoreaBio). Samsung Biologics launched a new CDO platform called S-Tensify, bolstering CDO competitiveness. S-Tensify supports high-intensity biomedicine development with the latest cell-culture technology. The company shared that S-Tensify’s adoption of N-1 perfusion technology has increased its cell-culture concentration by 30-fold, significantly boosting manufacturing capacity during inoculation at the seed stage (N-1). ST Pharm has started CDMO of CRISPR/Casx, used in gene editing technology. Gene editing is a biomolecular tool for editing precise locations within DNA using zinc finger nucleases, TALEN, or CRISPR/Cas9. CRISPR/Casx is an innovative technology for precisely editing DNA sequences, enabling deletion, addition, or correction of genetic material. Novel drug development using gene editing is currently underway. The U.S. FDA approved exa-cel (U.K.-authorized product name: Casgevy), a CRISPR/Cas9 gene-edited therapy developed by the United States biotech company Vertex Pharmaceuticals and SWISS-based CRISPR Therapeutics. ST Pharm showcased its manufacturing process of sgRNA, which precisely targets the genome. More than 100 mer high-purity sgRNA production is needed to develop medicines, and this manufacturing technology is more difficult than ASO or siRNA oligonucleotides ingredients. During the event, Prestige Biologics shared new CDMO business opportunities due to the imminent passing of the Biosecure Act by the United States. According to the company, over 30 companies have requested meetings, and 6 offers have been received. Prestige Biologics emphasized that the company is equipped with cost competitiveness and quality for a single-use approach. K-bio showcased novel drug development competitiveness In addition to CDMO orders, Korean biopharmaceutical companies also showcased their capabilities in the development of novel drugs. VaxCell Biotherapeutics presented the outcomes of Phase 2a ‘Vax-NK/HCC’ clinical trials for hepatocellular carcinoma. This trial evaluated the efficacy of Vax-NK/HCC plus HAIC combination therapy involving 16 patients with advanced hepatocellular carcinoma who did not respond to conventional treatments. The clinical results demonstrated that Vax-NK/HCC plus HAIC combination therapy had an objective response rate (ORR) of 68.8% and a progression-free survival (PFS) of 16.8 months. Genome & Company has confirmed the competitiveness of its ADC candidate. Earlier this month, the company signed a technology transfer contract with Swiss-based Debiopharm for its ADC candidate, GENA-111. Including the upfront payment of approximately KRW 6.9 billion, the contract size amounts to a maximum of KRW 586.4 billion. GENA-11 is an ADC candidate with a novel mechanism for targeting CD239. CD239 is known to be highly expressed in cancer cells than in healthy cells, and no ADC has been commercialized for targeting this. Genome & Company is considering targeting GENA-111 to treat gynecologic cancer. After assessing which payload would be more effective, the company stated that the final target indication would be decided. Bridge Biotherapeutics shared directives for developing novel drug candidates to treat idiopathic pulmonary fibrosis (IPF). BBT-887 is an innovative new candidate under development in a phase 2 trial. It is a selective inhibitor of autotaxine enzyme. Autotaxine is a protein known to bind to receptors in cells and induce various physiological activities, such as sclerosis and tumorigenesis. Bridge Biotherapeutics presentation (photo=Bridge Biotherapeutics). Last month, Bridge Biotherapeutics received authorization from the Independent Data Monitoring Committee (IDMC) to continue the clinical trial. After evaluating the efficacy and safety data of 75 clinical subjects, there were no concerns related to the drug safety or effects.
- Product
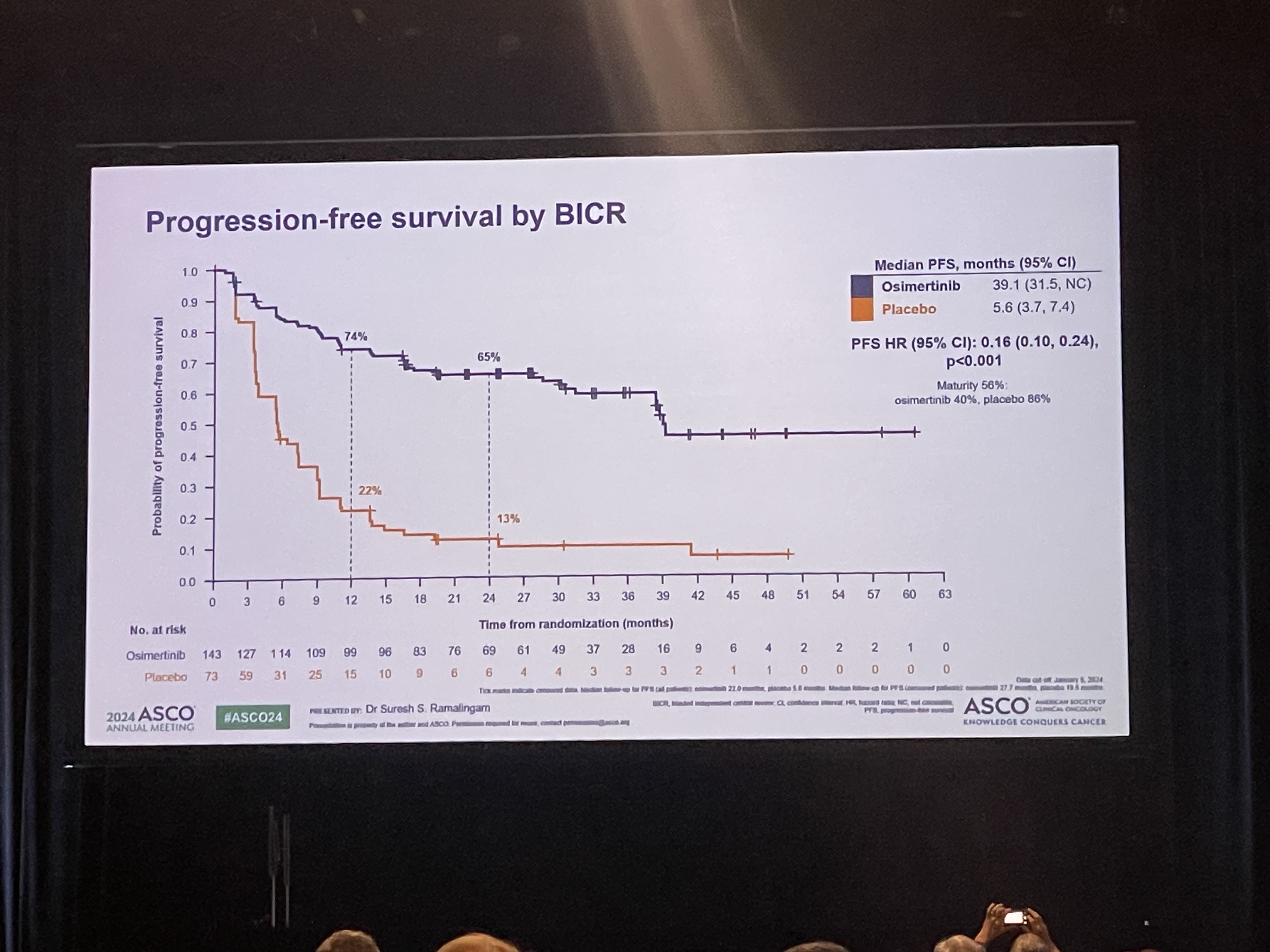
- Will Osimertinib emerge as the standard of care
- by Park, sang-jun Jun 10, 2024 05:41am
- The LAURA trial, which evaluated osimertinib’s effect in patients with unresectable Stage III EGFR-mutant non-small cell lung cancer who received chemoradiotherapy (CRT), was presented at the 2024 American Society of Clinical Oncology (ASCO) Annual Meeting. The results were also concurrently published in NEJM. # The LAURA trial evaluated progression-free survival (PFS) in 216 patients with unresectable stage III EGFR-mutant NSCLC who received chemoradiotherapy (CRT). The patients were randomized to receive osimertinib or placebo. The results showed a median PFS of 39.1 months and 5.6 months in the osimertinib and placebo arms, respectively, with an 84% reduction in the risk of disease progression and death in the osimertinib arm. The overwhelming numbers were met with spontaneous resounding ovation. Although the overall survival rates were not clear yet, researchers also added a positive interpretation based on the fact that overall survival did show a clear trend toward improved survival in the osimertinib arm, even though 80% of the placebo arm switched to osimertinib. Professor Suresh S. Ramalingam from the Winship Cancer Institute at Emory University School of Medicine, who presented results of the phase III LAURA study during the Plenary Session at the 2024 ASCO Annual Meeting, said, “The current standard of care for unresectable stage III EGFR-mutant NSCLC patients following CRT is durvalumab, but the benefit of the immunotherapy agent, specifically among patients with EGFR mutations, is uncertain. Based on the clear benefits, osimertinib after CRT will most likely emerge as the new standard of care for EGFR-mutant disease in this setting.” The next big question will be in setting the eligible subjects and timing of administration. Patients with unresectable stage III EGFR-mutant NSCLC who have received chemoradiotherapy (CRT) are regarded as an incurable group of patients, who have a high likelihood of relapse in the future. This is why drug use in this patient group needs to be reviewed from various aspects. Professor Lecia V. Sequist from the Massachusetts General Hospital and Harvard Medical School, who attended the presentation as a discussant for the abstract, regarded the results as a half glass of water, explaining that osimertinib may and may not be a viable treatment option depending on the perspective.” He emphasized that the positive benefits of osimertinib in terms of preventing brain metastases are a clear advantage, but the cost of the drug and increased side effects are a disadvantage. Professor Beung-Chul Ahn of the National Cancer Center, said, "The positive outcome of osimertinib in this group of patients is very welcome evidence, but if we evaluate it soberly, the cost of the drug cannot be ignored in clinical practice, and there are groups of patients who do not necessarily need it, so we need a treatment strategy that reviews its use according to the situation.”
- Company
- Chronic kidney disease drug ‘Kerendia’ can be prescribed
- by Eo, Yun-Ho Jun 10, 2024 05:41am
- Bayer’s new chronic kidney disease drug ‘Kerendia.’ ‘Kerendia,’ a treatment for chronic kidney disease, is now available for prescription after receiving approval for insurance reimbursement. According to industry sources, Bayer’s Kerendia (finerenone) has passed the drug committee (DC) of Big 5 tertiary general hospitals, including Seoul National University, Seoul Asan Hospital, and Sinchon Severance Hospital. Additionally, 49 hospitals in Korea have assigned Kerendia’s prescription code. Kerendia is a treatment for chronic kidney disease accompanying type 2 diabetes. It has been listed for reimbursement since February last year. With Chon Kun Dang becoming a marketing partner in South Korea, rapid landing and promotional activities are underway. Kerendia received approval in Korea last year. It is indicated for the treatment to reduce the risk of sustained eGFR (estimated Glomerular Filtration Rate, eGFR) decline, end-stage kidney disease, cardiovascular death, non-fatal myocardial infarction, and hospitalization for heart failure in adult patients with chronic kidney disease (CKD) associated with type 2 diabetes. CKD is one of the most common complications in type 2 diabetes and is an independent risk factor of cardiovascular diseases. While CKD is a progressive disease, it can be difficult to detect because the disease can progress without showing obvious signs until just before the late-stage renal failure occurs. With late-stage renal failure, patients require dialysis or kidney transplants to sustain life. This can pose a socio-economic burden and have a profound impact on a patient’s quality of life. For patients with type 2 diabetes, frequent monitoring and assessment of kidney damage and kidney function are essential. Early detection and appropriate treatment are critical to slowing down the progression of the disease and reducing the risk of cardiovascular diseases. In type 2 diabetes, the three key factors causing kidney disease are hemodynamic changes, metabolic abnormalities, and inflammation or fibrosis. However, in current therapy, treatments targeting hemodynamic and metabolic factors are only available, while treatments targeting inflammation and fibrosis are lacking, highlighting the need for new treatment approaches. Kerendia is a novel therapeutic approach targeting inflammation and fibrosis in adult chronic kidney disease patients with type 2 diabetes. It is the first non-steroidal, selective mineralocorticoid receptor antagonist. Overactivation of the mineralocorticoid receptor (MR) can lead to inflammation and fibrosis, which can result in permanent damages to kidney. Kerendia inhibits the overactivation of the mineralocorticoid receptor, reducing inflammation and fibrosis, and thereby preventing kidney damage. Kerendia demonstrated its effectiveness in the Phase 3 FIDELIO-DKD trials. FIDELIO-DKD trials enrolled approximately 5,700 patients from 48 countries globally, and Kerendia is indicated to inhibit the progression of chronic kidney disease and reduce the risk of cardiovascular events in adult patients with chronic kidney disease accompanying type 2 diabetes. Patients participating in the study received either Kerendia at doses of 10mg or 20mg in addition to standard therapy or a placebo. Clinical outcomes demonstrated that primary composite endpoint consisted of end-stage kidney disease, a sustained decline of more than 40% in eGFR, reduction by approximately 18% compared to the placebo in renal death. In addition, it reduced the secondary endpoints consisting of cardiovascular death, nonfatal myocardial infarction, stroke, or heart failure leading to hospitalization by approximately 14%. The outcomes of major adverse events or the rate of adverse events related to acute kidney damage were comparable between the two groups. Meanwhile, the European Society of Cardiology (ESC) revised its ‘2021 ESC Guidelines for the Diagnosis and Treatment of Acute or Chronic Heart Failure’ and listed Kerendia as a Class 1A recommendation to prevent hospitalization due to heart failure in patients with chronic kidney disease accompanying type 2 diabetes. Additionally, the ESC recommended annual assessment of eGFR and urinary albumin levels to screen for the development of chronic kidney disease in patients with diabetes.
- Company
- Pharma company MRs illegally work for CSOs on the sideline
- by Lee, Seok-Jun Jun 10, 2024 05:41am
- Contract Sales Organizations (CSOs) are trending in the pharmaceutical industry. In the case of small and medium-sized pharmaceutical companies, the companies have been opting to use CSOs rather than their own sales departments, to the extent that their departments are disappearing with the expansion of CSO business. The smaller the company, the more it relies on CSOs. A growing number of companies are outsourcing 100% of their sales. Large companies are also dabbling with CSOs. In 2019, the Ministry of Health and Welfare reported that 45% of the 195 pharmaceutical companies surveyed used CSOs. However, the industry consensus is that the rate would exceed 70% if the pool is narrowed to small and medium-sized companies. With CSOs becoming more mainstream, acts of misconduct are also increasing. One representative example is the act of pharmaceutical company medical representatives (MRs) illegally working for CSOs. According to the industry, many MRs who work for one company are selling their competitors' products at the same time. It's not uncommon to see MRs actively selling their competitors' products that pay higher commissions than their own. The problem is exacerbated by the industry's implicit acceptance of such illegal behavior. There are numerous posts on online job information sites recommending MRs to work for CSOs on the sideline, guaranteeing absolute confidentiality. There are even rumors that some small and medium-sized pharmaceutical companies that are in urgent need of making sales may tacitly allow employees to work for CSOs as long as they meet their sales targets. The CSOs are also encouraging the multicareer path. In fact, one CSO said, "The company can only see the wage and salary income it paid to each employee, so even if the employee earns additional income and reports it to the National Tax Service, the company will never know because it is categorized as business income. We provide full personal protection, so you don't have to worry about getting caught working a second job." Another CSO said, "We guarantee the highest commission in the industry. We work directly with pharmaceutical companies, so we can get the highest commissions. Our CSO is not like the others that are actually a bunch of corporate entities. No one knows how many steps the electronic data interchange (EDI) goes through in those companies, and whether there are any problematic entities, so it's really important to use a CSO that directly deals with the pharmaceutical company." The practice of pharmaceutical MRs doubling as CSOs needs to be abolished, as it can lead to a lack of collegiality and professional ethics, and can cause serious damage to the company in the form of trade secret theft and unfair competition. The industry is also aware of this problem and is making efforts to eradicate the practice. Companies are increasing penalties for MRs who are illegally working two jobs and expanding education and monitoring to prevent such acts. The CSO Reporting System is one of the most notable industry efforts. The CSO Reporting System is one of the key components of the amendments to the Pharmaceutical Affairs Act, which will come into full effect on October 19th. At its core, the bill requires "CSOs to report their business activities to local governments where they are located.” Violators will be punished by imprisonment for up to 3 years or a fine of up to KRW 30 million. Pharmaceutical companies will also be required to check their ability to prepare, keep, submit, and disclose expenditure reports, manage the appropriateness and transparency of their accounting, and be obligated to provide training to prevent illegal acts. The government believes that the introduction of the reporting system will be able to address the illegal practices by CSOs and pharmaceutical companies that circumvent the system, which has been raised as a possible source of illegal rebates. "At a time when the pharmaceutical industry as a whole is making efforts to integrate CSOs into the system, by disclosing expenditure reports and implementing the CSO reporting system, it is imperative that we eradicate and prevent illegal CSO business activities to restore the CSOs’ credibility in the domestic pharmaceutical industry.” Meanwhile, the general business structure of CSOs is as follows. The CSO is referred to as a "corporate CSO," and its employees are referred to as "dealers.” Dealers are usually sole proprietors who work for the corporate CSO, and the corporate CSOs work like middlemen, signing contracts with pharmaceutical companies and setting commissions that they later pay out to their dealers. The corporate CSOs recruit dealers, sign contracts with pharmaceutical companies, receive commissions for sales and redistribute them back to individual dealers.

- Policy
- Roche applies for Ocrevus' reimbursement in KOR
- by Lee, Tak-Sun Jun 10, 2024 05:41am
- Roche Korea applied for reimbursement of 'Ocrevus Inj. (ocrelizumab, Roche Korea),’ its multiple sclerosis treatment that was approved last month, to the Health Insurance Review and Assessment Service. The drug is administered twice a year, which is considered to have dramatically improved the dosing convenience for MS patients. The drug is a megablockbuster drug with global sales of about KRW 9 trillion in 2022. Celltrion is developing a biosimilar version of the drug. According to industry sources, Roche Korea submitted a drug determination application to HIRA for Ocrevus Inj on the 5th. Ocrevus Inj is a recombinant humanized monoclonal antibody (mAb, IgG1) that selectively targets CD20-expressing B cells, reducing the number and function of B cells to inhibit MS. MS is a disease of the central nervous system, which consists of the brain, spinal cord, and optic nerve, and is an autoimmune disease characterized by the patient's immune system attacking healthy cells and tissues. It is estimated that there are about 1,800 MS patients in Korea. The initial symptom is usually unilateral visual neuritis and is often accompanied by other various symptoms depending on the part of the central nervous system affected. Most patients complain of fatigue. Although there is no cure, various medications are being used to alleviate the symptoms. Beta interferon injections are the most commonly used, but the regimen requires receiving an injection from every other day to at least once a week. Ocrevus Inj offers greater dosing convenience with an initial dose of 600 mg administered in 2 intravenous infusions, followed by a single dose of 600 mg injection every 6 months. Ocrevus was approved by the U.S. FDA in March 2017 for the treatment of adult patients with relapsing or primary progressive forms of multiple sclerosis. In Korea, the Ministry of Food and Drug Safety approved the drug as a treatment for ▲ relapsing forms of multiple sclerosis (RMS) in adults, including clinically independent syndrome, relapsing-remitting multiple sclerosis, and active secondary progressive multiple sclerosis, and ▲ primary progressive multiple sclerosis (PPMS) in adults. In the U.S., the annual cost of the drug is USD 71,087, which is equivalent to KRW 97.73 million in Korea. Due to its high cost, how the company and government will share the reimbursement cost is likely to be an issue during reimbursement review. Ocrevus Inj generated global sales of KRW 9 trillion by 2022, leading Roche's growth. It is the No. 1 product in the multiple sclerosis treatment market. Celltrion submitted a Phase III IND for an Ocrevus Inj biosimilar to the European Medicines Agency (EMA) in May last year. Recently, the Korea Pharmaceutical and Bio-Pharma Manufacturers Association named Roche as the pharmaceutical company that will generate top global sales in 2028, citing data from global pharmaceutical market research institution Evaluate. Its products, the immuno-oncology drug Tecentriq, macular degeneration drug Vabysmo, and Ocrevus are expected to contribute to the high sales.
- Opinion
- [Reporter’s View] Finances limit reimbursement of drugs
- by Eo, Yun-Ho Jun 07, 2024 05:51am
- Insurance reimbursement standards and indications for a drug can differ. This is because the government’s pockets are not infinite under the National Health Insurance System. This is why there are always complaints in the field. Not all complaints can be resolved but there are some that evidently require resolution, that were made for incomprehensible “financial” reasons that are difficult to understand. Restrictions on age, duration, and switching drugs are typical examples. In the case of the age restriction, the standard is simple. For the same condition, infants, young children, or elderly patients may be excluded from coverage, or only certain age groups may be included due to concern about financial exhaustion. In this case, reimbursement is often extended later through reimbursement expansion measures. However, some drugs remain inaccessible for the patients. Restrictions set on the duration of use are a little different. Generally, reimbursement standards limit a drug's dosing interval based on the drug's clinical studies or authoritative international guidelines. However, there are also cases where reimbursement is restricted for "financial" reasons without any specific grounds for limiting the dosing interval. Restrictions on switching are the most frequent. Autoimmune diseases are the next big thing in the pharmaceutical industry after anticancer drugs, and numerous classes of drugs and same-class drugs with the same mechanism of action compete in the market. In the case of these drugs, the government often does not allow patients to be reimbursed for the first drug if they switch to another, meaning that if a patient is given a new drug with the expectation that it will work better than the first drug, but experience worse prognosis, he or she cannot go back to the old drug. The same situation was applied to drugs that were introduced long ago, and it took a long time before switching was granted reimbursement. In Korea, drug reimbursement has a significant impact on prescribing practices. Even if a patient needs a drug, doctors will often refuse to prescribe it if it is not reimbursed. This is why restrictions for financial reasons can be detrimental in fields where prescribing is essential. We ask the health authorities to trust the field’s judgment a little more.
- Company
- Immuno-oncology drugs demonstrate additional benefit at ASCO
- by Son, Hyung-Min Jun 07, 2024 05:50am
- Global pharmaceutical companies have demonstrated their immuno-oncology drugs’ effect in refractory solid tumors. Clinical results from major immuno-oncology drugs, including Imfinzi, Opdivo, and Yervoy, were presented at the 2024 American Society of Clinical Oncology (ASCO) Annual Meeting, which kicked off last month in Chicago, U.S. In the case of Imfinzi, the trial confirmed a survival benefit in small cell lung cancer. Imfinzi has shown promise in gastrointestinal cancers, including biliary tract and liver cancer. Such results paved the way for the company to add new indications. The combination of Opdivo and Yervoy improved survival in the first-line treatment of liver cancer. The combination has demonstrated efficacy against Nexavar and Lenvima in the first-line treatment of liver cancer, setting the stage for new competition. Imfinzi improves OS, PFS in small-cell lung cancer AstraZeneca's Imfinzi showed efficacy in small-cell lung cancer, an area with a high unmet need. The Phase III ADRIATIC trial evaluated the efficacy of Imfinzi monotherapy and the Imfinzi+Imjudo combination versus placebo in 730 patients with limited-stage small-cell lung cancer whose disease had not progressed after concurrent chemoradiation therapy (cCRT). ADRIATIC study design, which evaluated the efficacy of immuno-oncology drug Imfinzi (Source: ASCO 2024 lecture capture). Patients were randomized to receive a fixed dose of 1,500 mg of Imfinzi every 4 weeks alone or in combination with 75 mg of Imjudo, followed by Imfinzi every 4 weeks for up to 24 months. The primary endpoints were Imfinzi monotherapy’s progression-free survival (PFS) and overall survival (OS) vs placebo. Results showed that Imfinzi monotherapy reduced the risk of death by 27% compared with placebo. Median estimated OS was 55.9 months in the Imfinzi arm compared with 33.4 months in the placebo arm. Approximately 57% of patients in the Imfinzi arm were alive at 3 years, compared with 48% in the placebo arm, and Imfinzi therapy reduced the risk of disease progression or death by 24% compared with placebo. Median progression-free survival (mPFS) at 2 years was 16.6 months in the Imfinzi arm and 9.2 months in the placebo arm, with 46% of patients in the Imfinzi arm experiencing no disease progression at 2 years compared with 34% in the placebo arm. In addition to the small number of patients, small-cell lung cancer has a significantly lower number of treatments available than other cancers. According to a fact sheet published by the Korean Association for Lung Cancer (KALC) and the Korean Central Cancer Registry (KCCR) based on 2015 data, only about 13% of all lung cancer patients (2,658) were diagnosed with small cell lung cancer. As the first immuno-oncology drug to demonstrate efficacy in limited-stage small-cell lung cancer, whether Imfinzi can rise to become a new treatment option in the field is gaining attention. Opdivo+Yervoy improves survival as first-line treatment in liver cancer BMS announced results from the Phase III CheckMate-9DW trial, which evaluated the efficacy of the combination of Opdivo, a PD-1-targeted immuno-oncology drug, in combination with Yervoy, a CTLA-4-targeted immuno-oncology drug. Opdivo and Yervoy demonstrated an effect as first-line treatment in liver cancer (Source: ASCO 2024 lecture capture. BMS is exploring the possibility of securing multiple indications with the Yervoy plus Opdivo combination. The company is currently exploring the potential of the combination in liver cancer, metastatic colorectal cancer, squamous cell carcinoma, and head and neck cancer. The results presented at the 2024 ASCO Annual Meeting are from an interim analysis of the trial that evaluated the efficacy and safety of the combination as a first-line treatment for liver cancer. The trial enrolled 668 treatment-naive adults with unresectable hepatocellular carcinoma who were randomized to receive the Opdivo+Yovoy combination and either Lenvima or Nexavar. Results showed a median OS of 23.7 months in the Opdivo+Yervoy combination arm, compared with the 20.6 months in the control arm. The objective response rate (ORR) was 36% with the Opdivo+Yervoy compared with 13% in the control arm. The median duration of response was 30.4 months for the Opdivo+Yovoy combination arm compared with 12.9 months for the control arm. These results lay the foundation for the Opdivo+Yovoy combination to become a significant competitor in the first-line liver cancer treatment market. Currently, the combinations Tecentriq (immuno-oncology drug) plus Avastin (targeted therapy) by Roche and Imfinzi (immuno-oncology drug)+Imjudo (targeted therapy) by AstraZeneca are available as first-line treatment for liver cancer. In the field, HLB's targeted anti-cancer drug rivoceranib in combination with Hangseo Pharmaceutical's immuno-oncology drug camrelizumab has also recently presented data showing improved survival.