- LOGIN
- MemberShip
- 2026-05-02 14:55:42
- Product
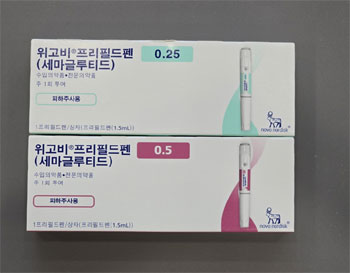
- Consumers use tricks to receive Wegovy prescriptions
- by Kang, Hye-Kyung Oct 22, 2024 05:51am
- With heating interest in the obesity drug Wegovy, the way-around measures the consumers are taking to receive prescriptions are causing controversy. From how to receive Wegovy through non-face-to-face treatment venues without the legwork to how to split the high-dose formulations being shared online, concerns are growing over the drug’s misuse. The first issue is non-qualified prescriptions. Prescriptions that ignore the prescribing criteria, such as obese patients with a body mass index (BMI) of 30 kg/m2 or more, are becoming widespread, and one of the outlets for such is the non-face-to-face treatment platforms. As influencers with tens of thousands to hundreds of thousands of followers are revealing the fact that they have been prescribed Wegovy, non-face-to-face treatment platforms such as ‘Dr Now’ and ‘My Doctor’ are being mentioned as venues for such prescriptions. You can receive a prescription for up to 5 pens of Wegovy through non-face-to-face treatment, and can also compare drug prices at pharmacies. The reason for choosing non-face-to-face treatment is that it is easier to receive prescriptions and dispense compared to face-to-face treatment. Although some prescribing medical institutions have been promoting Wegovy prescription through press releases, blogs, and social media including Instagram, hospitals and pharmacies have not yet been able to secure enough supplies, which is why patients are turning their attention to non-face-to-face treatment, which is relatively easier to receive prescriptions and dispensing. These platforms allow users to check the information, contact details, and prices of Wegovy in pharmacies with stock at a glance, making it easy to receive Wegovy without having to make many phone calls. Looking at the non-face-to-face treatment platforms, the price of Wegovy prescriptions by pen ranges from KRW 5,000 for 1 pen, KRW 7500 for 2 pens, KRW 10,000 for 3 pens, and KRW 10,000 to KRW 15,000 for 4 pens. For dispensing, a pharmacy in Gwangju had the lowest price in the country at KRW 419,000. In addition, some blogs and other websites have been sharing advice on how to get prescriptions easily, with tips such as ‘raising your weight.’ Another problem is the indiscriminate spread of information on its off-label use, such as ‘how to use Wegovy at half the price’. The correct way to start from the 0.25mg dose, then increase to 0.5mg, 1.0mg, 1.7mg, and 2.4mg over a four-week titration period, but many people have been prescribed 2.4mg and have been sharing tips such as splitting the dose into multiple doses. In fact, one doctor described it as ‘using the same drug at half the price’ and said, ‘I just use a 2.4mg syringe from the start. Then you can use a single syringe for four 0.25mg doses = 1/4 syringe, and four 0.5mg doses = 1/2 syringe, and so on.’ The argument is that since the price per dose is the same, it's more cost-effective to take one pen of 0.25mg over 4 doses than one pen of 0.5mg over 8 doses. It's also easier to stock the higher doses than the starter doses of 0.25mg and 0.5mg. Pharmacists are wary that their fears are being realized. Pharmacist A pointed out that “non-face-to-face treatment is promoting medical shopping in conjunction with the Wegovy craze. Shouldn't there be sanctions against random prescriptions and off-label use that are being conducted with non-face-to-face treatment?” Pharmacist B also said, “I am concerned that the Wegovy craze is spreading like a fad on social media without any warnings or side effects.” In particular, regarding dividing the high-dose formulation, B added, “Wegovy can be stored for up to 6 weeks after opening. If you split the dose, you'll be forced to take it beyond that period. I don't think I can give such advice from a professional perspective.” ‘The role of the government, pharmaceutical companies, and healthcare professionals will be crucial in tackling this Wegovy craze,’ he added. The Korea Pharmaceutical Association also said it plans to respond strongly to concerns such as the abuse of non-face-to-face Wegovy prescriptions and the delivery by documenting cases. Meanwhile, Novo Nordisk has warned patients in its patient instructions not to use Wegovy Prefilled Pen if they are hypersensitive (allergic) to semaglutide or any of the drug's excipients, or if they are pregnant, nursing mothers, etc., and emphasized patients to refrain from using the drug unless indicated.
- Company
- Will combination therapies for cancer be reimbursed in KOR?
- by Whang, byung-woo Oct 21, 2024 05:49am
- As the government has begun to prepare the principles for the reimbursement review of combination therapies that use new anticancer drugs, attention is being paid to whether the discussion will progress further. According to industry sources, the Health Insurance Review and Assessment Service recently held a Cancer Disease Deliberation Committee meeting and decided to prepare deliberation principles to discuss whether to approve benefits for major combination therapies. The decision is in line with the growing calls for a change in the reimbursement paradigm for prescribing anticancer drugs based on accumulated evidence on ‘combination therapies’ between treatments from various clinical studies. Recently, multinational pharmaceutical companies have been seeking the reimbursement of combination therapies using new anticancer drugs, calling for an institutionalized process. When considering reimbursement for combinations of non-reimbursed new drugs and reimbursed chemotherapy drugs, the industry has suggested that discussions on previously reimbursed drugs should be reserved and only the non-reimbursed drugs should be discussed. A case in point is AstraZeneca's immuno-oncology drug Imfinzi (durvalumab), the reimbursement of which had been discussed for biliary tract cancer last year. At the time, CDDC only approved gemcitabine and cisplatin in combination with chemotherapy (the GemCis regimen) for first-line treatment of biliary tract cancer, while leaving Imfinzi non-reimbursed. The government will discuss setting a deliberation principle where a drug, which is already reimbursed, is used with a non-reimbursed new drug as part of combination therapy and should continue to be reimbursed without further discussions. This can be observed in the results of the 7th CDDC Review, which reviewed the combination of ‘endocrine therapy’ and ‘Verzenio (abemaciclib)’ for the treatment of early breast cancer. The outcome of the deliberations was to keep the previously reimbursed adjuvant endocrine therapy as is and make the newly added Verzenio available on a ‘100% coinsurance’ basis, reducing the cost burden on the patients. In other words, it means that the process will be simplified by allowing the reimbursement of existing treatments within the combination to be recognized. However, these principles are not automatically applicable to all new combination therapies, and as it leaves the existing framework in place, it is far from the improvements to the new drug approval approaches the industry had hoped for. Nevertheless, it is positive as it has opened the door to further discussions. There are also expectations that such discussions will lead to solutions for ‘new drug+new drug’ combination therapies. A pharmaceutical industry official said, “It is still a drug-by-drug approach, so it's not a one-size-fits-all approach, but it's positive that the discussions have been officially made. We understand that KRPIA and others are also having discussions on what kind of processes will be required for the reimbursement of new drugs for combination therapies in the future,’ “With the number of combination therapy cases increasing, there is currently no set process for how applications should be submitted for listing and how they should be reviewed. We are looking forward to the various approaches that may be developed through discussions as individual cases have come to light.”
- Policy
- 2nd GIFT drug Nefecon’s approval imminent in Korea
- by Lee, Hye-Kyung Oct 21, 2024 05:48am
- Meditip's Nefecon (budesonide), which was designated as the 2nd Global Innovative Products on Fast Track (GIFT) drug last year, is close to receiving approval in Korea. According to industry sources on the 21st, the Ministry of Food and Drug Safety recently completed the safety and efficacy review for Nefecon. The completion of the safety and efficacy review in general leads to approval in Korea. Nefecon has been designated as a GIFT item as a drug that developed a new efficacy and effectiveness using the already marketed budesonide ingredient. A GIFT designation can also be given to a previously approved ingredient that has no treatment for a disease but can be used to treat a new patient population. Nefecon is a drug used to treat primary IgA nephropathy in adults with a urinary protein-to-creatinine ratio of 1.5 or greater who are at risk of rapid progression. In a presentation made at Kidney International, Nefecon was associated with a 27% lower urinary protein-to-creatinine ratio at 9 months of treatment compared to placebo. Glomerular filtration rate remained stable, with a difference of 3.87 ml/min/1.73 m2 compared to placebo. The drug was approved by the US FDA last year under the brand name Tarpeyo and by the European EMA under the trade name Kinpeygo. In China, the National Medical Products Administration (NMPA) designated Nefecon as a Breakthrough Therapy Designation (BTD) in 2020, and the Taiwan Food and Drug Administration granted it an Accelerated Drug Designation (ADD). IgA nephropathy is a disease caused by the deposition of immune complexes, including IgA, in the glomeruli of the kidneys, causing an inflammatory response. About 9,000 patients are known to be affected by IgA nephropathy in Korea. In clinical practice, antihypertensive drugs such as ARBs and ACEIs, immunosuppressants, and diuretics are used to treat IgA nephropathy. However, these drugs are symptomatic treatments that prevent the worsening of symptoms, and no drug fundamentally treats the condition. Meanwhile, drugs are given the GIFT designation if it is a ▲ drug aimed at treating serious diseases such as life-threatening cancer or rare diseases ▲ drug aimed at preventing or treating infectious diseases that may cause serious harm to public health, such as bioterrorism infectious diseases or pandemics of infectious diseases ▲ new drug developed by a Korea Innovative Pharmaceutical Company as designated by the Ministry of Health and Welfare ▲ a combination of a drug and medical device subject to fast-track review ▲ a case where there is no existing treatment or a drug showed clinically significant improvement in efficacy over existing treatments. The MFDS reviews the eligibility for the GIFT designation when a pharmaceutical company applies for expedited review and, if necessary, designates a drug as a GIFT item after consulting with the Medical Product Expedited Review Expert Council. Once designated, the review period is reduced by at least 25% (e.g. from 120 working days to 90 working days). Applicants will receive a variety of support for rapid commercialization, including rolling review, where the application data is reviewed on a rolling basis, close communication between the reviewer and the developer, such as item briefings and supplementary briefings, and expert consulting on regulatory matters.
- Company
- Therapeutic device 'improves OS for lung cancer patients'
- by Son, Hyung Min Oct 21, 2024 05:48am
- The first therapeutic device has been approved for use in non-small cell lung cancer (NSCLC). Novocure's therapeutic device, in combination with an NSCLC medication, demonstrated to improve patients' overall survival and won the approval of the U.S. regulatory authority. In South Korea, Nu Eyne is researching the potential of oncology therapeutic devices through clinical studies. According to industry sources on October 19th, the U.S. Food and Drug Administration (FDA) granted approval for Novocure's Optune Lua for the treatment of metastatic non-small cell lung cancer (NSCLC). Optune Lua is approved for use in combination with PD-1/PD-L1 immunotherapy for cancer or docetaxel. Optune Lua is a device that delivers 'Tumor Treating Fields (TTFields),' which exert physical forces on the electrically charged components of dividing cancer cells, resulting in cell death. Novocure says that TTFields do not affect healthy cells because they have different properties (including division characteristics, morphology, and electrical properties). The basis of approval was the Phase 3 LUNAR study. The clinical trial involved 301 patients with NSCLC who had failed during or after platinum-based chemotherapy. The patients were randomly assigned in a 1:1 ratio to the group receiving Optune Lua plus a PD-1/PD-L1 inhibitor or docetaxel or to the single-treatment group receiving a PD-1/PD-L1 inhibitor or docetaxel. Clinical results showed Optune Lua combination therapy recorded a median overall survival (OS) of 13.2 months, set as the primary endpoint. This was 3 months longer than 9.9 months in the control group. Patients randomly administered Optune Lua plus immunotherapy had a median OS of 19.0 months, and patients administered a PD-1/PD-L1 inhibitor alone had a median OS of 10.8 months. The Optune Lua combination therapy group had an OS extension of over 8 months compared to patients treated with immunotherapy alone. Patients randomly administered Optune Lua plus docetaxel had a median OS of 11.1 months, whereas patients treated with docetaxel had a median OS of 8.9 months. However, this figure did not show a statistically significant difference. Device-related adverse events (AE) occurred in 63.1% of patients treated with Optune Lua combination therapy. Most cases were Grade 1-2 and only 4% (6 patients) experienced Grade 3 skin toxicity and discontinued treatment. Optune Lua did not show Grade 4-5 toxicity, and no deaths occurred. Therapeutic device is being developed in South Korea…Nu Eyne challenges the field A Korean company, Nu Eyne, has confirmed the effects of immunotherapy and is conducting therapeutic device development. To confirm treatment effects on cancer, Nu Eyne conducted a cell-based clinical study to compare the stimulated group to the non-stimulated group of cultured cells after 48 hours of electrical stimulation. The results showed that the electrically stimulated group had an up to 80% reduction in tumor size. Nu Eyne's technology for its oncology therapeutic device has been developed to deliver a high-intensity electrical field that responds particularly to cancer cells, thereby inducing cell death. The technology is known to have a low risk of causing side effects on surrounding organs. Nu Eyne is developing advanced systems and materials to maximize oncology therapeutic effects. Using its technology, Nu Eyne aims to develop therapeutic devices to secure indications for treating lung cancer and bone metastatic cancer. Nu Eyne aims to complete the development of a clinical therapeutic device prototype in the second half of 2026. After that, the company plans to conduct clinical trials to secure the FDA regulatory approval.
- Company
- Oxlumo receives orphan drug designation in Korea
- by Eo, Yun-Ho Oct 21, 2024 05:48am
- The primary hyperoxaluria treatment Oxlumo received orphan drug designation in Korea. The Ministry of Food and Drug Safety (MFDS) recently announced the designation through an orphan drug designation notice. Oxlumo (lumasiran) was also recently designated a Global Innovative products on Fast Track (GIFT) by the MFDS. The drug is an RNAi therapeutic for primary hyperoxaluria (PH1), a rare kidney disease that was approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) in 2020. RNAi is recognized as a next-generation gene therapy that has the advantage of providing specific access to disease-causing human genes. PH1 is a rare disease caused by excessive production of oxalate in the liver. Symptoms include the deposition of oxalate crystals or potassium oxalate crystals in the kidneys and urinary tract. As the disease progresses, kidney damage occurs and dialysis is required. Eventually, a liver or kidney transplant is required to cure the disease. The option to treat PH1 with a therapeutic agent became available with the approval of Oxlumo in 2020. Oxlumo is an RNAi therapy that targets hydroxy acid oxidase 1 (HAO1), which encodes glycolate oxidase (GO), the enzyme that produces oxalate. The mechanism of action reduces oxalate by inhibiting HAO1, which reduces GO production. Oxlumo’s efficacy was confirmed through a Phase III clinical trial in 39 PH1 patients aged 6 years and older. Oxlumo reduced oxalate levels in the urine by 65.4% compared to the placebo group. In addition, 84% of patients treated with Oxlumo had urinary oxalate levels near normal. 52% achieved urinary oxalate levels within the normal range.
- Company
- RNAi therapeutic 'Givlaari' receives the ODD in KOR
- by Eo, Yun-Ho Oct 21, 2024 05:48am
- Product photo of Givlaari (givosiran) RNAi therapeutic 'Givlaari' has been designated as an orphan drug following its designation as the GIFT. The Ministry of Food and Drug Safety (MFDS) recently announced this through the posting of the Orphan Drug Designation (ODD). Givlaari (givosiran) had previously been designated as the 'Global Innovative products on Fast Track (GIFT)' by the MFDS. This drug won U.S. FDA approval in 2019 and also received the accelerated approval from Europe's EMA. Givlaari is a subcutaneous injection that targets the enzyme aminolevulinic acid synthase 1 (ALAS1) for the treatment of acute hepatic porphyria (AHP) in adults and adolescents aged 12 years and older. AHP is caused by a genetic deficiency due to the depletion of the enzyme needed for heme biosynthesis in the liver. It is a rare genetic disorder with abnormal accumulation of porphyrins in the body. Patients with AHP experience debilitating symptoms, such as severe abdominal pain, vomiting, and seizures, and chronic pain. The efficacy of Givlaari has been demonstrated through the Phase 3 ENVISION clinical trial. Givosiran was shown to reduce the occurrence of annual porphyria-associated seizures by 74% compared to the placebo. The research outcome was published in The New England Journal of Medicine (NEJM) on June 11th, 2020. During the six months of treatment, the percentage of patients who had not experienced porphyria-associated seizures was 50% for the givosiran-treatment group and 16.3% for the placebo group.
- Policy
- How to stabilize supply of national essential medicines
- by Lee, Hye-Kyung Oct 18, 2024 05:49am
- While prescribing drugs by ingredient name and introducing international nonproprietary names (INNs) are being discussed as measures to stabilize the supply of drugs, voices have emerged calling for a system that allows the use of the same ingredient drugs as nationally essential medicines. The issue of stabilizing the supply of not only national essential medicine but also drugs that are constantly experiencing supply and demand instability is one of the most frequent questions asked by the National Assembly's Health and Welfare Committee's national audit this year. According to the written inquiries submitted by the MFDS to the National Assembly on the 17th, 7 lawmakers - Yoon Kim, Hee-Seung Park, Jong-Heon Baek, Mi-Hwa Seo, Young-Seok Seo, Byung-Hoon So, and In-soon Nam - inquired about stabilizing the supply of medicines. Representative Yoon Kim pointed out the unstable supply of national essential medicines and asked how the MFDS plans to promote the use of drugs with the same ingredient name. “Changing the method of prescriptions is the responsibility of the Ministry of Health and Welfare,” said the MFDS, adding, ’The MFDS will cooperate with the competent ministry, which is MOHW, if requested.” For national essential drugs, the Stable Supply Consultative Group is discussing comprehensive measures, improving the criteria for designating pediatric drugs as national essential medicines and the designation and removal national essential medicines. In particular, in the case of pediatric drugs, products that are essential for pediatric patients but have unstable supply are designated as national essential medicines. The MFDS said, “In the case of medicines whose supply is expected to be interrupted (shortage) due to insufficient production, we will jointly respond with the Ministry of Health and Welfare through the Public-Private Consultative body for Unstable Supply and Demand of Medicines, reviewing measures including raising drug prices.’ Representative Hee-Seung Park inquired about the reasons for the supply chain instability of national essential drugs, to which the MFDS responded, “We understand that the supply chain instability of national essential drugs is intensifying due to a combination of factors such as increased demand due to infectious diseases such as COVID-19 and deterioration of solvency due to rising production costs.” There was also an inquiry about the supply interruption report, which is required to be reported to the MFDS 60 days before the supply interruption. On this, the MFDS announced on the 4th of this month that the ‘Rules on the Safety of Drugs, Etc.’ will be amended to change the supply interruption reporting criteria from 60 days to 180 days in advance from April 5th next year. Until now, reporting drug shortages had been a recommended measure, but from next year, it will become mandatory. “In order to promptly receive drug supply interruption reports, the deadline for reporting supply interruptions has been moved up from 60 days to 180 days in advance,” said the ministry, adding, “If a pharmaceutical company has a plan to reduce production and imports below a certain standard, it must file a report within 1 month.” Representative Jong-Heon Baek called for a stable supply plan through the expansion of domestically produced drugs. “We support domestic pharmaceutical companies to develop manufacturing process technologies for national essential medicines that are highly dependent on imports and require stable supply and demand, and will continue to strive for the stable supply and self-sufficiency of raw materials and finished products for national essential medicines that are essential for healthcare,” the MFDS explained. Through the self-sufficiency of national essential medicines project, which has been underway since 2022 with the Korea Orphan & Essential Drug Center and the Korea Pharmaceutical and Bio-Pharma Manufacturers Association, the associations completed the development of 3 raw materials (amiodarone, ketoconazole, and benserazide) and 2 finished drugs (amiodarone injection and tablets) in the first phase (2022-2023). As the second phase (2024~2026) of the project, the company is developing 3 raw materials (acetaminophen, ipratropium, and furosemide) and 2 finished products (acetaminophen and furosemide). The MFDS also responded to Representative Young-Seok Seo’s inquiry about the need to monitor the process of distributing medicines that are difficult to supply to pharmacies and medical institutions. The MFDS said, “The MOHW is in charge of reporting on the supply of medicines, and the two ministries are responding to timely identification of supply and demand issues through smooth information exchange. We will continue such information exchange in the future so that the relevant ministries can jointly respond to drug supply insecurity.” It was also pointed out that compared to direct response measures such as emergency import or made-to-order production, the measures made by the ministries end at indirect measures such as monitoring and encouraging production. “There are many reasons for drug supply interruptions, such as production issues and raw material supply problems, and we are responding to each cause by reviewing the existence of substitute drugs and consulting with experts,” the MFDS said, adding, “We will prepare a response manual for drug supply interruptions in the future.”
- Policy
- 'Can the Yoon administration develop blockbuster drugs?'
- by Lee, Jeong-Hwan Oct 18, 2024 05:49am
- Criticism has been raised that the government's policy to create global blockbuster drugs is not working and has a dark future. It has been pointed out that the difficulty in raising funds to support the R&D costs for pharmaceutical and biotech companies and ventures is causing financial difficulties for bioventures, and that expert human resource training is not being done properly, either. On the 17th, lawmaker In-soon Nam of the Democratic Party of Korea questioned Soon-do Cha, President of the Korea Health Industry Development Institute, about the policy direction of the pharmaceutical bio-industry during the National Assembly audit of the agency. Rep Nam said, “At the beginning, there was some talk that Cha is closely communicating with President Suk-Yeol Yoon. I'm not pointing any fingers, but I was expecting the business of the KHIDI to progress better.” ‘The Yoon administration is saying that we should support the development of the pharmaceutical bio industry as a future growth engine, but not much speed nor progress is being made in the field. The administration said that it will support the creation of 2 blockbuster drugs by 2027, but can it really achieve this?” “We have set an implementation plan and are checking the implementation of the comprehensive plan every year,” Cha replied, “I know it is difficult, and there are no guarantees, but we are proceeding surely and cautiously.” Nam criticized Cha's tendency to give generalized answers and requested more specific result reports. “What I'm hearing is that biotech companies are facing financial difficulties,” Nam said, adding, “New investments should be flowing in, but they're decreasing. I wonder if the Yoon administration is really setting a policy direction for the pharma-bio industry as Korea’s future food.” “I heard that the pharma-bio fund has not been created yet. Are they fostering human resources? Please submit your progress in detail before the comprehensive audit,” said Nam.

- Company
- Bladder cancer drug Balversa offers new treatment option
- by Whang, byung-woo Oct 18, 2024 05:49am
- The introduction of the targeted therapy Balversa (erdafitinib) in urothelial carcinoma has attracted attention for its potential to address unmet needs. In particular, the emergence of FGFR inhibitors has highlighted the importance of genetic mutation diagnostics to quickly detect the presence of such mutations. (from the left) Professor Tae-Jung Kim, Professor Inho Kim, BU director Yeon-Hee Kim Inho Kim, professor of Oncology at Seoul St. Mary's Hospital, and Tae-Jung Kim, professor of pathology at Yeouido St. Mary's Hospital, emphasized the importance of diagnosing FGFR mutations at a press conference held by Janssen Korea on the 16th, which was held to celebrate the launch of Balversa. Balversa was approved by the Ministry of Food and Drug Safety in January 2022. However, it is still not reimbursed in Korea. Specifically, the drug is indicated for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma (mUC) with FGFR2 or FGFR3 genetic alterations whose disease has progressed on or after at least one line of prior systemic therapy, which includes platinum-based chemotherapy, or whose disease has progressed within 12 months of neoadjuvant or adjuvant treatment with platinum-based chemotherapy. However, the approval of PD-1 and PD-L1-directed immuno-oncology agents in the first- and second-line settings that followed Balversa’s approval led to the need for Balversa to demonstrate efficacy in patients who previously received these agents. The situation was addressed with the publication of Balversa’s Phase III THOR trial study, which demonstrated a prolonged overall survival (OS) benefit with Balversa over chemotherapy in patients with metastatic urothelial carcinoma with FGFR3/2 gene alterations whose disease progressed after first-line treatment with immuno-oncology agents. In the study, Balversa prolonged overall survival (OS) compared with chemotherapy in patients with metastatic urothelial carcinoma. Results showed that over a median follow-up of 15.9 months, the mOS was 12.1 months in the Balversa arm, reducing the risk of death by 36% compared with the 7.8 months in the chemotherapy arm. Based on these findings, the U.S. Food and Drug Administration granted Balversa formal approval in January, but with a more restricted indication than originally approved. “Bladder cancer is most commonly diagnosed in those in their 60s and older, with frequent recurrences and metastases, so it is important to prevent metastases or treat recurrences and metastases early,” said Professor In-ho Kim. “There is a significant unmet therapeutic need, especially for patients with distant metastases, where the 5-year relative survival rate is only 11.7%.” ‘Balversa is the first targeted therapy for bladder cancer, which is significant because it improves survival in patients who have exhausted both chemotherapy and immuno-oncology options and provides an opportunity for further treatment,’ added Professor Kim. The second speaker, Professor Tae-Jung Kim, emphasized the importance of early diagnosis of FGFR mutations in bladder cancer patients. ‘FGFRs play a significant role in signaling pathways that regulate cell growth, differentiation, survival and migration,’ said Professor Kim. ’FGFR mutations are found in a variety of cancers, but they are particularly common in bladder cancer, where they are observed in approximately 20% of patients.’ ‘The use of mutation-specific targeted therapies may help stop cancer proliferation and progression or improve the effectiveness of other treatments,’ he added. ’The NCCN guidelines also consider or recommend molecular/genomic testing for genetic mutations in some patients, such as those with bladder cancer tumor invasion grade IIIB or higher.’ In other words, for patients with bladder cancer who are in the chemotherapy phase, the guidelines recommend testing for genetic mutations in the early stages of treatment strategy planning. “We are pleased to be able to offer a new treatment option in FGFR-mutated urothelial cancer with Balversa. We plan to communicate the clinical benefits and emphasize the importance of mutation diagnosis so that more patients can benefit from Balversa treatment.”
- Company
- 'Vorasidenib' receives Orphan Drug Designation in KOR
- by Eo, Yun-Ho Oct 18, 2024 05:49am
- 'Vorasidenib,' an anticancer drug targeting brain cancer, has been designated an orphan drug in South Korea. The Ministry of Food and Drug Safety (MFDS) announced this on October 8th through the Orphan Drug Designation (ODD) notice. Vorasidenib is an orally administered dual inhibitor of isocitrate dehydrogenase (IDH) 1/2 that selectively penetrates the blood-brain barrier. The drug's efficacy for selective malignant brain tumors (neuroglioma) was demonstrated through a double-blind Phase 3 clinical trial led by a research team at the University of California, Los Angeles (UCLA). The research team administered the targeted anticancer drug vorasidenib (40 mg single dose per day) or placebo to 331 patients with specified brain tumors (residual or recurrent Grade 2 glioma with IDH mutation) who have undergone brain tumor surgery as their only treatment. Grade 2 gliomas with isocitrate dehydrogenase (IDH) mutation are malignant brain tumors that cause considerable disability in patients, leading to early death. Gliomas tend to advance slowly but are fatal and affect people in their 30s. Study participants were randomly assigned to vorasidenib-treated patients (study group) and placebo-treated patients (control group). 168 of these patients took vorasidenib, and 163 took the placebo. Study results showed that the progression-free survival (PFS) for the vorasidenib-treated patient group was 27.7 months and that for the placebo-treated patient group was 11.1 months. Vorasidenib delayed the progression of malignant tumors by 16.6 months and lowered the death risk to 39%. Additionally, it improved the time until the next anticancer therapy (chemotherapy, radiotherapy), reducing the patient death risk to 26%. The side effects experienced by the vorasidenib-treated patient group were 22.8%, whereas 13.5% in the placebo-treated patient group. However, no significant side effects have been found. The primary endpoint was imaging based PFS and the secondary endpoint was the time to the next anticancer intervention. Of those study participants, 226 (about 68%) patients had a follow-up of 14.2 months (median value) and continued to take vorasidenib or placebo. At 30 months (Sept. 2022), 72% of the vorasidenib-treated patient group was still taking the drug and had not experienced disease progression. Meanwhile, France-based Servier Pharmaceuticals developed vorasidenib and is proceeding with the approval process in the U.S. and Europe.