- LOGIN
- MemberShip
- 2026-05-02 21:37:52
- Policy
- Novartis cuts price of Cosentyx after PVA negotiations
- by Lee, Tak-Sun Nov 07, 2024 05:46am
- Cosentyx UnoReady Pen The price of Novartis’s Cosentyx(secukinumab) is set to be reduced due to its increased use. This is because the National Health Insurance Service recently reached an agreement during Price-Volume Agreement negotiations with the company for Cosentyx. According to industry sources on the 6th, the insurance price ceiling of Cosentyx was adjusted for the second time under the PVA system after 2021. The affected products are the Cosentyx SensoReady Pen and Cosentyx UnoReady Pen. Cosentyx was listed for reimbursement in Korea in August 2017. Currently reimbursed indications include ▲chronic severe plaque psoriasis, ▲active and progressive psoriatic arthritis, and ▲severe active ankylosing spondylitis. According to the market research institution IQVIA, Cosentyx SensorReady Pen has seen an increase in sales every year since its reimbursement. In 2019, sales reached KRW 12.3 billion, then KRW 19.9 billion in 2020, KRW 25.1 billion in 2021, KRW 30.3 billion in 2022, and KRW 31.7 billion in 2023. In addition, Cosentyx UnoReady Pen, a high-dose product that was launched last year, generated sales of KRW 4.5 billion. Due to such high demand, the insurance price ceiling of Cosentyx had been adjusted in 2021 under the PVA system. At the time, the price of Cosentyx Sensoready Pen dropped by 7.3% from KRW 682,938 to KRW 630,084. At the time, 'Type A’ of the PVA negotiations was applied. Type A negotiations are applied to listed new drugs whose claims in the same product category have increased by more than 30% from the expected claims amount. In 2020, the drug had already well exceeded the expected claims amount. Three years have passed, and this year, ‘Type B’ PVA negotiations were applied. Type B negotiations are applied when the claims for the same product group, whose insurance price ceiling had been adjusted by Type A negotiations, or four years passed after the date of initial listing or the date the price ceiling was adjusted through negotiations, have increased by 60% or more than the previous year's claims, or by 10% or more but the increase is more than KRW 5 billion. Since Cosentyx had its price ceiling adjusted through Type A negotiations, it is possible that the additional negotiation for price adjustments was ordered because its claims increased by more than 60% or more than KRW 5 billion over the previous year's claims. Given that Cosentyx Sensoready’s sales grew 5% YoY in 2023 by KRW 1.4 billion (IQVIA), the introduction of Cosentyx UnoReady Pen, which falls in the same product group, may have triggered the PVA negotiation. Combined, the increase in sales amounted to more than KRW 5 billion compared to the previous year. Upon the agreement, the final price of Cosentyx is expected to be announced after deliberation by the Ministry of Health and Welfare’s Health Insurance Policy Review Committee.
- Policy
- 'Eutropin·Biktarvy' price drop amid sales hike
- by Lee, Tak-Sun Nov 06, 2024 05:52am
- Product photos of Eutropin and Biktarvy. Due to the increased volume of usage, drug pricing for 'Eutropin Inj (somatropin, LG Chem)' and 'Biktarvy (Bictegravir/Emtricitabine/Tenofovir Alafenamide, Gilead Science),' which recently showed a sales hike, is expected to be reduced. Sources said on November 5th that the Health Insurance Review and Assessment Service (HIRA) recently agreed on drug pricing adjustment of these products through the 'Type-Na' negotiation. As a result, the final adjusted prices will be listed on the reimbursement list following a review from the Health Insurance Policy Review Committee. Eutropin has been a top-selling growth hormone drug in the Korean market. 'Eutropin S Pen,' introduced in April 2022, is skyrocketing in sales. Eutropin S Pen's expiration date has been extended from 18 months to 24 months, and, based on UBIST, it generated sales of KRW 81.5 billion last year. Eutropin recorded KRW 21.3 billion in sales in the same period. The combined sales growth rate for the two products is 24% compared to last year. Biktarvy also successfully achieved two-digit sales figures. Based on IQVIA last year, it generated sales of KRW 54.5 billion, up 11% from the previous year (KRW 49.1 billion). Biktarvy received the MFDS approval in January 2019, and it has been in sales following reimbursement listing in July. It is a single-capsule combination drug for the treatment of HIV containing three active ingredients: Bictegravir, Emtricitabine, and Tenofovir alafenamide. With Yuhan and Gilead Sciences in a joint-sales agreement, Biktarvy is a top-selling drug in the HIV market. "Eutropin and Biktarvy show rapid growth in the market," an expert in the pharmaceutical industry said. "With the increased usage volume, reducing its ceiling price according to the Price-Volume Agreement (PVA) is inevitable." In 2022, the ceiling prices of Eutropin and Biktarvy were lowered due to the PVA agreement. Eutropin was reduced by 2.9%, and Biktarvy was reduced by 3.0%. Meanwhile, the negotiation of the PVA 'Type-Na' is conducted when the ceiling price is adjusted by the type-Ga price or when the bill amount of the same product has increased by more than 60% or 10% from the previous year's bill, and the increase is more than KRW 5 billion.

- Company
- 20-valent pneumococcal conjugate vaccine set to launch
- by Moon, sung-ho Nov 06, 2024 05:52am
- Attention has been drawn to another competitor set to launch in the market for the 'pneumococcal vaccine.' This year, the pneumococcal vaccine market has been fiercely competitive with newly released products. As the 20-valent vaccine is likely to come out right after the 15-valent conjugate vaccine has entered the market after 13 years, competition between global pharmaceutical companies to take the market share is becoming intense. As clinical practices draw attention to the next generation of vaccines, they expect that 'price' will be a determining factor in the non-reimbursed market. According to pharmaceutical industry sources, the Ministry of Food and Drug Safety (MFDS) granted approval of Pfizer Korea's 'Prevenar 20 Prefilled Syringe (hereafter, Prevenar 20).' Prevenar 20 is Pfizer Korea's next-generation product that follows 'Prevenar 13,' which currently dominates the Korean market for pneumococcal vaccines. In addition to serotype components in the 13-valent vaccine, Prevenar 20 contains seven additional serotypes, including 8, 10A, 11A, 12F, 15B, 22F, and 33F. The MFDS has approved the drug's indication to prevent invasive pneumococcal disease, pneumonia, and acute otitis media caused by pneumococcus in infants, children, and adolescents aged six weeks and below 18 years old. It is also used to prevent invasive pneumococcal disease and pneumonia caused by pneumococcus in adults 18 years of age and above. As Prevenar 20 has been officially approved, Pfizer Korea will prepare to launch the product. Pfizer Korea will likely aim to launch the product in the first half of 2025. Analysis suggests that an official launch is possible when Prevenar 20 is included in the vaccine guidelines of major medical sciences organizations and the National Immunization Program (NIP). The process is similar to that of MSD's 15-valent vaccine, 'Vaxneuvance,' which has been officially launched into the clinical practices. MSD Korea received the official approval of Vaxneuvance from the MFDS on October 31, 2023. Considering Pfizer Korea's Prevenar 20 was approved on October 31, 2024, Vaxneuvance was approved precisely a year before. After the approval, Vaxneuvance was included in the Korean Society of Infectious Diseases (KSID)'s '2024 Updated Guidelines for Adult Immunization' that "KSID recommends use of the 15-valent pneumococcal conjugate vaccine (PCV15, MSD's Vaxneuvance) over the 13-valent pneumococcal conjugate vaccine (PCV13, Pfizer's Prevenar 13) for individuals subjected to adult pneumococcal conjugate vaccine (PCV) immunization." MSD launched the vaccine in April this year soon after its inclusion in the NIP, and Pfizer Korea is likely to follow a similar launching schedule. "We have been trying to receive Prevenar 20's approval. The specific schedule for launch has not been decided," a representative from Pfizer Korea said. "We will prepare for the launch next year." "We have yet to confirm our Korean partner for Prevenar 20," a representative from Pfizer Korea said. "We are in a partnership with Chong Kung Dang for Prevenar 13 for adults, and we have an agreement with 'Koreavaccine' for Prevenar 13 for young children." The industry draws attention to the competition in the pneumococcal vaccine market for the clinical practice. The question is who will dominate the market, given the competition between two global pharmaceutical companies. Following the withdrawal of the 10-valent PCV vaccine (Synflorix, GSK) from the Korean market, the 13-valent vaccine Prevenar 13 and the 15-valent vaccine Vaxneuvance are currently competing in the clinical practice. In particular, MSD Korea chose Boryung Biopharma as its Korean partner. Upon launching Vaxneuvance, MSD Korea actively engaged in sales‧marketing by hiring Paik Jong Won, a businessman, TV celebrity, and CEO of TheBorn Korea, as its commercial model. According to a pharmaceutical market research firm UBIST, the number of NIP and non-reimbursed immunizations for adults in nationwide hospital‧private clinics is rising after Vaxneuvance launched in April. In detail, based on UBIST, the cumulative number of PCV vaccine immunizations from April to June this year is 994 counts. Among these, 809 immunizations were with Prevenar 13. 185 Vaxneuvance immunization was made. After MSD Korea launched Vaxneuvance in April this year, they chose Paik Jong Won, TheBorn Korea CEO, as their commercial model, putting efforts into raising awareness. To the right is the product photo of Prevenar 20, Pfizer Korea's pneumococcal vaccine, which is expected to launch in the first half of 2025. The number of immunizations with Prevenar 20 is below that of market dominating Prevenar 13, but the number is rising. Based on the assumption that Vaxneuvance has been well established in clinical practice as a new vaccine, the number of immunizations were likely increased. In fact, a director of the pediatrics and adolescent clinic said, "When we used Prevenar 13 for immunizations, we had several inquiries if it's possible to switch to Vaxneuvance. Since medication switching is possible during the immunization course, we answer patient inquiries." And added, "If patients have not completed the immunization schedule, medication switching is possible. We had several cases of switching after inquiries." Considering changes brought by the Vaxneuvance launch this year, Prevenar 20 will likely bring changes to clinical practices such as the department of internal medicine and the department of pediatrics and adolescents. Sang Hyuk Ma, a Director of Pediatrics and Adolescents at Changwon Fatima Hospital, said, "Following Prevenar 13, Vaxneuvance and the 15-valent vaccine have been introduced to South Korea. We must consider clinically if a vaccine with more serotypes is appropriate for South Korea. Pneumococcal serotypes tend not to mix, and serotypes common in South Korea must be accounted for vaccination." Ma added, "In other words, the vaccines were developed towards what's needed in the United States. We must look into common serotypes in South Korea and discuss whether the latest vaccines are necessary for immunizations in Korean citizens, including infants and children." Gwak Kyung-Keun, the President of Seoul Physicians' Association, said, "In my opinion, there is no difference between the 15-valent and the 20-valent vaccines. There is no basis for which one is more superior." Gwak anticipated, "In the end, it will be the sales‧marketing competition between companies. One would argue that the 20-valent vaccine has more serotypes but will be a marketing fight without a comparison basis."

- Company
- Roche seeks digital solutions for its healthcare ecosystem
- by Whang, byung-woo Nov 06, 2024 05:52am
- With the importance of personalized healthcare being emphasized more than ever with the emergence of innovative new drugs, Roche Diagnostics Korea is aiming to lead the market by focusing on digital solutions. The company plans to lead the healthcare sector through digital transformation in line with the paradigm shift in treatment, from early diagnosis and precision medicine to predictive medicine. (From the left) Tae-Hyun Um (Director of Insurance Policy Affairs, KSLM), Yeo-Min Yun, Director of Scientific Affairs, KSLM), Sail Chun (CEO&Chaiarman, KSLM), Kit Tang (General Manager of Roche Diagnostics Korea), Muhwan Yun (head of Digital Insights, Roche Diagnostics Korea), Sungho Cho (Head of Core Lab & POC, Roche Diagnostics Korea) On the 5th, Roche Diagnostics Korea held a press conference on the theme of “Future Healthcare and Innovation Presented by Diagnostic Tests” and emphasized the importance and opportunities of digital transformation in the healthcare sector. With the launch of the Digital Insights division, the company has been focusing on providing data-driven insights and marking a new turning point in personalized medicine. “Digital transformation in healthcare is the new normal, and we have already entered the era of artificial intelligence (AI) transformation beyond digital transformation,” said Muhwan Yun, head of Digital Insights at Roche Diagnostics Korea. ”Roche Diagnostics has set its own ethical standards for applying AI to healthcare, and is aiming to expand and innovate its digital portfolio, including continuous R&D investment and collaboration.” One representative project is Smart Lab, a digital transformation of diagnostic laboratories. The goal is to make existing laboratory operations more efficient, flexible, and data secure, to ultimately increase insight from analyzing data. For this, Roche Diagnostics has been emphasizing 'digital solutions' and building an enabling ecosystem. It focuses on incorporating significant data of a certain scale, advanced analytics, and digital technologies to support medical decisions and improve the treatment experience. The company’s representative technology, NAVIFY, is a cloud-based data platform that analyzes vast amounts of healthcare data in a standardized format to enable precision medicine. “In practice, we have seen significant improvements in time, manpower, and cost metrics for testing and analysis after implementing Roche Diagnostics Digital Insight Solution’s NAVIFY portfolio,” said Yun, ”and the satisfaction rate among healthcare providers who have used them is over 90%.” “With South Korea set to enter a super-aged society next year, the role of diagnostic test data for efficient and effective treatment is expected to increase further,” said Kit Tang, General Manager of Roche Diagnostics Korea. ”Roche Diagnostics Korea will continue to strive to contribute to an efficient healthcare system and improved patient outcomes with innovative diagnostic solutions covering a wide range of disease areas.” The hurdle to building a digital ecosystem for diagnostic tests is in the “maturity” of the digital transformation In the long run, digital transformation in diagnostics is essential, but there are hurdles. Not only are institutional improvements required for the digital transformation of the existing system, but the nature of healthcare is such that it is important to integrate the different perspectives of each stakeholder, including hospitals, manufacturers, patients, and academic societies. Yeo-Min Yun (Director of Scientific Affairs, KSLM)In response to this, Yeo-Min Yun, Director of Scientific Affairs at KSLM of the Korean Society for Laboratory Medicine (Konkuk University Hospital), emphasized that the emergence of technology should be supported by measures that can encourage their use in practice, such as by assigning service fees. “If you look at imaging, the technology that analyzes the images through AI technology is being applied and utilized to create values that have never existed before,” said Director Yun. ”Referring to this precedent, I think that efforts should be made to apply a service fee for such diagnostic tests and recognize their value in clinical practice.” Ultimately, Roche Diagnostics' challenge is to collect and utilize fragmented data. To this end, the company expects the newly launched division to serve as a base to build the ecosystem. “In Korea, data compatibility is relatively poor, so we are striving to collect and utilize data well,” said Director Yun, adding, ”As the implementation of digital ecosystems is not fully mature yet, it will be important for each component, including the company, to be integrated.”
- Company
- CKD’s CKD-508 receives approval to initiate P1T in the U.S.
- by Son, Hyung Min Nov 06, 2024 05:52am
- Chong Kun Dang announced on the 4th that it has received approval from the U.S. Food and Drug Administration (FDA) for the Phase I clinical trial of CKD-508, its new drug candidate for dyslipidemia. In the trial, Chong Kun Dang will confirm the safety and lipid-improving effects of CKD-508 and explore the optimal dose for a Phase II trial. CKD-508 is a treatment for dyslipidemia that works by inhibiting the activity of cholesteryl ester transfer protein (CETP), which facilitates the transport of cholesteryl esters (CE) and triglycerides (TG) between lipoproteins in the blood, thereby lowering low-density cholesterol (LDL-C) levels and increasing high-density cholesterol (HDL-C) levels. In a non-clinical efficacy trial conducted by Hyosung Research Center, Chong Kun Dang confirmed the LDL-C-lowering and HDL-C-raising effects of CKD-508 and demonstrated a significant reduction in apolipoprotein (Apo-B), a key marker of dyslipidemia. “CKD-508 is an innovative drug that is expected to be effective at low doses by solving the problems of previous CETP inhibitors that were discontinued due to drug accumulation and blood pressure increase based on its strong binding to CETP,” said Chong Kun Dang. ”If successful, it is expected to become a new treatment option for patients with statin-resistant dyslipidemia that cannot be controlled with statins.”
- Policy
- How will the new drug review process change with fee hike?
- by Lee, Hye-Kyung Nov 06, 2024 05:52am
- The Ministry of Food and Drug Safety (MFDS) announced plans to raise the fee for new drug approvals to KRW 410 million from January 1 next year. The agency has prepared the approval and review process to implement the plan and has begun collecting industry opinion. According to MFDS’s press corp coverage, the MFDS recently delivered a revised version of the “Operating Procedures for Approval and Examination of New Drug Products (Guideline for public officials)” to the pharmaceutical and biotech industries and relevant associations. The guidelines contain specific measures to implement the “Innovative Measures for New Drug Approval” that was announced by the MFDS on Sept. 9, including the operation of a dedicated review team for each product and prioritizing GMP-GCP on-site inspections. “Upon the announcement of the rise in the approval fee of new drugs under the Innovative Plan for New Drug Approval, the pharmaceutical industry raised the question of whether the actual approval period will be shortened,” said an MFDS official. ”By recruiting reviewers, GMP inspections will be prioritized to be completed within 90 days from the date of the application receipt, and pre-registration of supplementary materials and explanatory meetings will reduce unnecessary delays in the approval process, dramatically shortening the approval period for new drugs from 420 days to 295 days.” The enactment of the Guideline on Operating Procedures for New Drug Approval and Review was promoted in line with the redetermination of new drug approval fees, which will take effect on January 1, 2025, and includes various changes to differentiate it from the existing approval process, such as the formation of a dedicated team, the establishment of explanatory meetings, and the introduction of a procedure for pre-registration of supplementary materials. From next year, when a new drug approval application is filed, MFDS will conduct an independent preliminary review within 7 days. During the preliminary review period, 80% of the fee will be refundable, and the MFDS will check the submission requirements. If there are any missing materials during this process, the applicant will have 14 days to submit them before an “initiation meeting” is held. Given the overall review timeline, the preliminary review process is crucial as data not submitted by the initiation meeting will be difficult to submit later and the application will be categorized as an application that requires primary supplementary data. A key part of this process is the establishment of a dedicated team for each item before the initiation meeting. The head of the approval division will lead the team, and a representative from the approval division will take charge as each item’s manager, and the team will be composed of representatives from ▲safety, efficacy, ▲quality control, ▲GMP (good manufacturing practice), and ▲GCP (good clinical practice). In addition, an initiation meeting is held within 14 days of receipt to coordinate the entire approval and review process and to provide the applicant with the necessary materials and procedures. The initiation meeting may be held face-to-face, via video, or in parallel, which is expected to increase the efficiency of the new drug review process. According to the proposed Operating Procedures for Approval and Examination of New Drug Products, GMP inspections will be conducted within 90 days from the date of application receipt, and GCP inspections will be conducted within 60 days after the first supplement. By accelerating the inspection schedule compared to the existing procedure, the verification required for the manufacturing and clinical management procedure will be completed at an earlier stage. This is intended to speed up the approval process and ensure adequate quality control. A number of improvements have also been made to the supplement requests. Previously, there were only first and second supplementary notifications, but the reform introduces a first supplementary request briefing session and a second supplementary request briefing session to ensure that the applicant fully understands the supplementary request. In particular, the explanatory meetings provide detailed information on the scope and requirements of the required materials and provide time to answer the applicant’s questions. In particular, for the first round of supplemental data submissions, a pre-registration process and a request for an information session can be made by the applicant. Applicants can pre-register their supplementary data and request the KFDA to hold an explanatory meeting. This allows the adequacy of the supplementary data to be reviewed in advance, reducing additional supplementary work and increasing the efficiency of the approval process. In addition, a final meeting will be held 5 days before the final approval target date to discuss the results of the approval review. “In addition to the effect of shortening the approval period, the revised procedure will improve the transparency of the approval and review process and help pharmaceutical and biotech companies develop new drugs efficiently while protecting the public's health,” said an MFDS official. ”We will collect opinions from relevant associations and industries to prepare the final draft and continue to optimize and implement the approval review service for the development of the domestic pharmaceutical industry.”
- Company
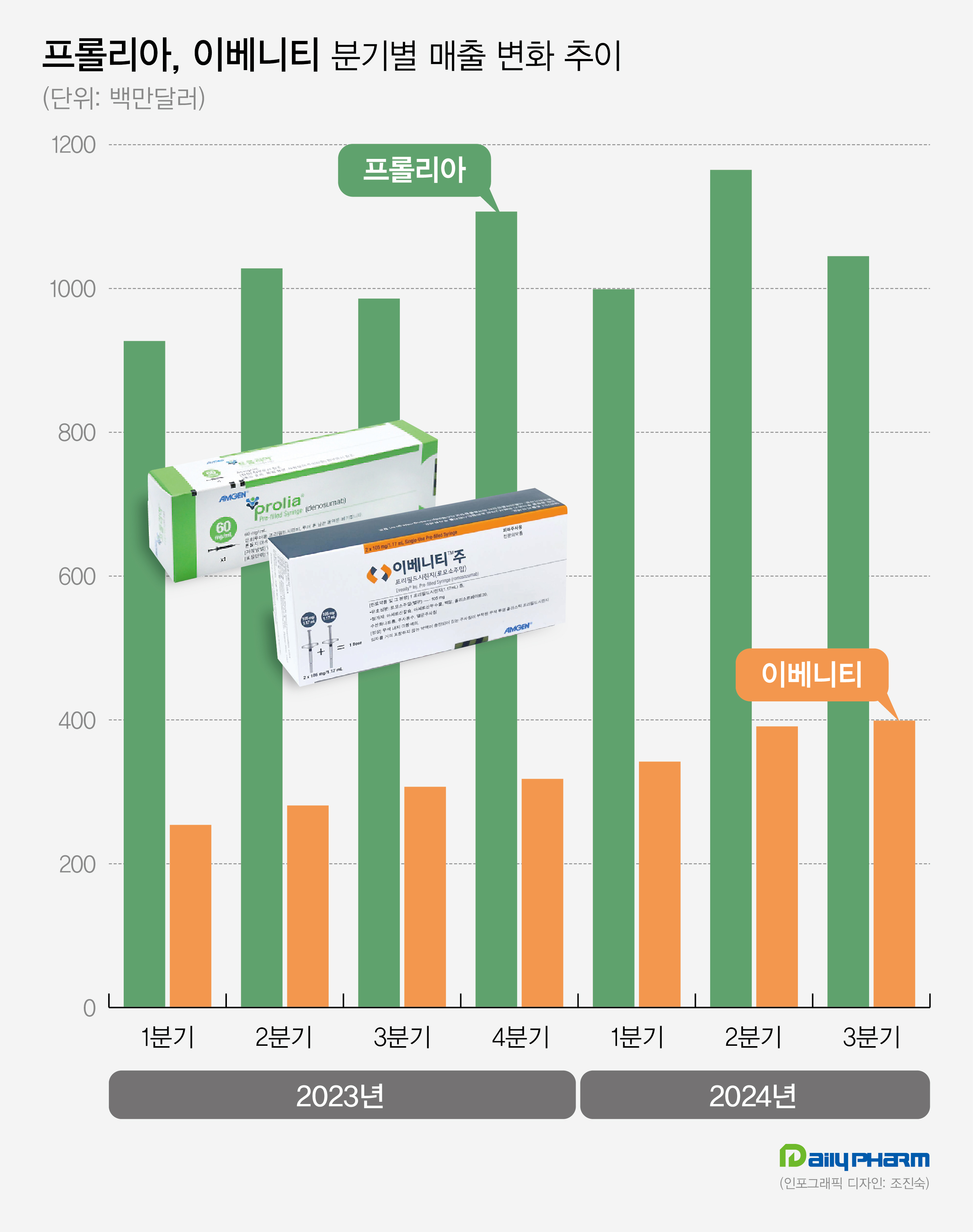
- Amgen's Prolia·Evenity generate KRW 2T in quarterly sales
- by Son, Hyung Min Nov 06, 2024 05:52am
- Amgen Amgen's Prolia and Evenity for the treatment of osteoporosis continue to show sales growth. Prolia and Evenity led Amgen's performance, generating sales of KRW 2 trillion in Q3. Analysis suggests that sequential therapy, using Evenity followed by Prolia for the treatment of osteoporosis with high-risk fractures, has been widely used, and it has led to continued increases in sales. Sources said on November 6th, Amgen's sales in Q3 were US$ 8.50 billion (approximately KRW 11.72 trillion), up 23.1% year-over-year (YoY). Operating profit for the same period was US$ 2.047 billion, up 1.3% from 2022. Prolia generated the highest sales among Amgen's products. Prolia's sales in Q3 were US$1.045 billion (approximately KRW 1.44 trillion), up 6.0% YoY. Prolia recorded US$3.20 billion (approximately KRW 4.42 billion) for the nine months this year, up 9.1%. Last year, Prolia's annual sales were US$40.48 billion. Developed by Amgen, Prolia is a treatment for osteoporosis. It was approved in the United States, and after that, Amgen successfully received Prolia's approval in major countries, including South Korea, Europe, and Japan. Prolia sales nearly doubled to US$352 million in Q1 of 2016 in two years from US$196 million (approximately KRW 270 billion) in Q1 of 2014. Prolia's sales continue to grow, generating US$852 million (approximately 1.2 trillion) in Q1 of 2022. In Q2 of 2023, its quarterly sales topped US$1 billion. In Q1 of this year, it dropped to US$9.99 billion, but then successfully rebounded in Q2, recording US$1.165 billion. Prolia's strength lies in having secured real-world data. Based on its FREEDOM and FREEDOM Extension clinical studies spanning 10 years, Prolia effectively reduced the risk of fractures than the conventional osteoporosis treatment, Alendronate. Unlike other medications, Prolia continues to increase bone density. Prolia is categorized as a medication that can be administered for long-term. As a result, pharmaceutical companies are developing Prolia biosimilars. Earlier this year, U.S.-based Sandoz received approval for a biosimilar of Prolia and Korean pharmaceutical companies such as Celltrion and Samsung Bioepis also plan to complete clinical trials and prepare for approval. Prolia's patent is set to expire in 2025. Quaterly sales performance of Prolia (green) and Evenity (oranage) in 2023 and 2024 (unit: US$1 million). Evenity records KRW 550 billion in Q3 sales…continues to increase The success of the osteoporosis treatment Evenity has contributed to Amgen's sales performance. Evenity's Q3 sales amounted to US$399 million (approximately KRW 550 billion), up 30.0% from US$307 million in 2022. Evenity is a bone-forming agent that provides dual effects of promoting bone formation and inhibiting bone absoption. It was approved in the United States in 2019. Evenity recorded sales of US$100 million for the first time in Q1 2020, and it continues to grow. In Q3 2022, Evenity recorded sales of US$200 million, more than doubling its initial launch. In Q3 of Last year, Evenity surpassed US$300 million and showed steady growth, reaching sales of US$400 million in Q3 of this year. Evenity's increase in sales is mainly due to its osteoporosis treatment strategy. As part of its marketing strategy, Amgen has strategized a sequential therapy of using the bone-forming agent Evenity followed by the bone resorption inhibitor, Prolia. Based on their approved indications, Bone-forming agents have a maximum duration of use of 2 years. Therefore, after a certain period of treatment, switching to bone resorption inhibitors such as Prolia or bisphosphonate agents are mainly used. According to Evenity's ARCH study or a study comparing teriparatide and risedronate, a sequential therapy using bone-forming agents first followed by bone resorption agents is more effective in preventing the risk of bone fractures in patients. Korea-based or overseas guidelines for osteoporosis treatment recommend bone-forming agents, such as Evenity, as first-line treatments for high-risk individuals.
- Company
- Chong Kun Dang speeds up dyslipidemia drug development
- by Son, Hyung Min Nov 05, 2024 05:46am
- Chong Kun Dang is accelerating the development of new drugs for dyslipidemia. The company has received approval to initiate a second global clinical trial in 10 years for its new drug candidate 'CKD-508' since the company began its research in 2014. According to industry sources on the 4th, Chong Kun Dang recently received approval from the U.S. Food and Drug Administration (FDA) to initiate a Phase 1 clinical trial on its dyslipidemia drug candidate, 'CKD-508'. Through the clinical trial, Chong Kun Dang plans to verify the safety and lipid-improving effect of CKD-508 and explore the optimal dose for its future Phase 2 trial. CKD-508 is a second-generation drug that overcomes the challenges of first-generation cholesteryl ester transfer protein (CETP) inhibitors, including limitations such as off-target effects (unexpected side effects caused by the drug), accumulation of fat cells, and poor stability. The drug candidate works by inhibiting the activity of CETP, which facilitates the transport of cholesteryl esters (CE) and triglycerides (TG) between fatty proteins in the blood, thereby lowering low-density lipoprotein cholesterol (LDL-C) levels and raising high-density cholesterol (HDL-C) levels. More than 6 years after initiating the CKD-508 study in 2014, Chong Kun Dang has now initiated global clinical trials. In June 2020, the company received approval from the UK's MHRA for a Phase 1 clinical trial of CKD-508 and is advancing its research and development. Preclinical results showed that CKD-508 significantly reduced LDL-C and LDL-C-containing apoprotein (Apo-B) and increased HDL-C in animal models with dyslipidemia. In addition, CKD-508 did not cause drug accumulation in adipose tissue or an increase in blood pressure, which were observed in clinical trials of anacetrapib and torcetrapib. Anacetrapib and torcetrapib were candidates for dyslipidemia developed by Merck and Pfizer, but the companies discontinued their development in 2017 due to safety concerns in the clinical trial stage. Chong Kun Dang believes that CKD-508 has the potential to be a breakthrough drug that could provide a new treatment option for patients with dyslipidemia that is not controlled by existing drugs such as statins. CKD-508 can be administered once weekly and may offer improved convenience over existing therapies. Along with CKD-508, Chong Kun Dang is also conducting 5 clinical trials in the synthetic drug category, including CKD-943 for uremic pruritus, CKD-950 (namodenoson) for hepatocellular carcinoma, CKD-951 for metabolic dysfunction-associated steatohepatitis (MASH), and CKD-510 for rare diseases. The company completed clinical trials for CKD-943 in the U.S. in 2015 and is currently in Phase III clinical trials. Chong Kun Dang has been developing CKD-943 since 2012 after signing a license agreement with Cara Therapeutics in the US for the exclusive development and sales of CKD-943 in Korea. CKD-950 recently entered Phase II clinical trials in Israel. In 2016, Chong Kun Dang entered into an exclusive domestic license agreement with Israel's Can-fite Biopharma for CKD-950, a novel liver cancer drug candidate. The company also acquired a domestic patent for CKD-950 in 2020. CKD-951, which is being developed as a treatment for MASH, has been recruiting patients since its IND approval in 2020. CKD-951 has a mechanism of action that improves inflammatory and fibrotic responses by acting on A3AR, which is overexpressed in liver inflammatory/fibrosis-promoting cells. CKD-510 was licensed out to Novartis in November of last year. CKD-510 is a histone deacetylase 6 (HDAC6) inhibitor that uses the company’s highly selective, non-hydroxamic acid platform technology. The drug candidate has demonstrated safety and tolerability in Phase I clinical trials in the U.S. and Europe.

- Company
- ‘Prevent MI recurrence through efficient LDL-C control'
- by Whang, byung-woo Nov 05, 2024 05:45am
- With the rise of metabolic diseases such as hypertension, diabetes, and hyperlipidemia increase in Korea, the prevalence of myocardial infarction and atherosclerotic cardiovascular diseases are also on the rise. The mortality rate of myocardial infarction is in the 20-30% range when it occurs for the first time, but the mortality rate increases sharply to 68-85% when it recurs, which is why efforts to prevent recurrence are being stressed now. In particular, one of the hot topics in treatment is how to manage LDL cholesterol, which is known to be an important factor in preventing the recurrence of atherosclerotic cardiovascular disease (ASCVD). In recent years, treatment options have become more diverse and multiple approaches have been proposed. Dr. Dong-Oh Kang, Professor of Cardiology and Cardiovascular Center at Korea University Guro Hospital, emphasized the need to effectively lower LDL cholesterol levels in high-risk patients. Dong-Oh Kang, Professor, Department of Cardiology, Korea University Guro Hospital “New drugs have changed the approach to LDL cholesterol management in high-risk patients” In severe cases of acute myocardial infarction, stenting or balloon angioplasty is performed to open up the blood vessel, as it is an emergency treatment for blocked blood vessels or low blood flow. However, these procedures are reactive, and it is important to use medications to prevent the same event from happening again. “It is important for patients who have had a myocardial infarction to use drugs to prevent further accumulation of atherosclerotic plaque and narrowing of the artery,” said Professor Kang. ”Lowering cholesterol to inhibit the progression of atherosclerotic plaque and preventing blood clots has become a key treatment.” This is why one of the most important topics in recent guidelines is to what level LDL cholesterol should be lowered in very-high-risk patients. Both domestic and international academic societies have proposed a strict management standard for patients with a history of atherosclerotic cardiovascular disease, with LDL cholesterol targets of less than 55 mg/dL and at least 50% lower than baseline. “The past guidelines suggested that LDL cholesterol levels could be as low as 100 mg/dL, but more potent drugs have come in a variety of combinations.” said Professor Kang, “As lowering LDL cholesterol levels has been shown to reduce the risk of atherosclerotic cardiovascular disease, even lower levels are now being recommended.” According to Kang, the suggested LDL cholesterol level for high-risk patients was less than 70 mg/dL in the 2010s, but by the late 2010s, patients with coronary artery disease or at very-high-risk were advised to lower their LDL cholesterol level to less than 55 mg/dL and at least 50% from baseline. In particular, the European guidelines suggest lowering LDL cholesterol levels to less than 40 mg/dL for patients with acute coronary syndrome who have had a recurrent event within the last 2 years. “Cardiologists who see patients with more severe acute myocardial infarction or patients undergoing procedures seem to be in agreement with the lower LDL cholesterol targets. However, some have concerns about lowering LDL cholesterol levels below 55 mg/dL or 70 mg/dL.” Diversification of treatment options, including PCSK9 inhibitors...“Strategy will change depending on reimbursement status” As Professor Kang noted, the lower LDL cholesterol target levels have been accompanied by the emergence of drugs that can effectively lower the levels to such targets. In the past, statins, which inhibit the synthesis of cholesterol in the liver, were the only drugs available to lower LDL cholesterol levels, but more strategies became available with the introduction of ezetimibe, which inhibits cholesterol absorption in the intestine, including statin and ezetimibe combinations. Then, the entry of monoclonal antibody drugs such as Repatha (evolocumab), a PCSK9 inhibitor, into the reimbursement system has transformed the clinical landscape. Currently, PCSK9 inhibitors are used in patients with myocardial infarction whose LDL cholesterol levels have not dropped sufficiently despite the use of high-intensity statins and ezetimibe. “It's important to monitor the dose escalation during initial therapy,” said Kang. “If LDL-C targets are not met, the dose should be increased and the patient reevaluated. If the maximum dose is not effective, a PCSK9 inhibitor such as Repatha, which has a faster LDL cholesterol lowering rate and is more potent, may be considered.” “In terms of Repatha’s effect, 19 out of 20 people will have lower LDL cholesterol level maintained, even at 30 mg/dL. In patients who had low LDL cholesterol, to begin with, we see reductions to less than 10 mg/dL.” In the long term, the introduction of oral bempedoic acid and injectable siRNA therapies is expected to further expand treatment options. In addition to access to treatments based on patient condition, Professor Kang predicts that treatment approaches will change based on the drug’s reimbursement status. “As more effective treatments will continue to be developed, we expect more and more combination options to emerge, and it is necessary to prescribe them considering the patient's condition and the characteristics of each drug,” said Kang. ”Since there are various drugs, their use will likely be determined by how reimbursement is applied in high-risk patients.” In addition to secondary prevention, Kang emphasized the need for policy promotion to screen and manage patients before they become high-risk. “Even though people are sufficiently screened and informed about their risk factors through health screenings, they often overlook them and look back in retrospect after they become ill. It is necessary to always receive screening and make efforts to properly treat or improve lifestyle habits from the primary prevention stage.”
- Company
- New oHCM drug 'Camzyos' nearing approval for reimb in KOR
- by Eo, Yun-Ho Nov 05, 2024 05:45am
- Product photo of Camzyos. 'Camzyos,' a new drug to treat obstructive hypertrophic cardiomyopathy (oHCM), is nearing 90% approval for insurance reimbursement listing. Sources said that BMS Pharmaceutical Korea and the National Health Insurance Service (NHIS) concluded drug pricing negotiations for Camzyos (mavacamten), a new drug for obstructive hypertrophic cardiomyopathy (oHCM). The drug had previously faced a delay in the decision, but the company quickly reached an agreement this time. As a result, Camzyos is likely to be listed within this year. This drug received a re-assessment status during the Drug Reimbursement Evaluation Committee (DREC) review of the Health Insurance Review and Assessment Service (HIRA). After that, it passed the DREC review and entered a drug pricing negotiation in August, but the drug did not receive a decision during the negotiation period (60 days). Camzyos is the only drug that selectively inhibits cardiac myosin-actin cross-bridge formation, which is the cause of oHCM. Camzyos' mechanism involves dissociating myosin from actin, relaxing overstimulated heart muscle, and thereby improving left ventricular outflow tract (LVOT) structure and LVOT outflow obstruction. Due to the lack of available treatments for oHCM for a long time, off-label medications have been used to manage symptoms. After Camzyos launched, the European Society of Cardiology (ESC) updated its guidelines for managing cardiomyopathy for the first time in about nine years. Previously, the guidelines for HCM were based on evidence limited to small-scale monitoring data, retrospective analysis results, and consensus opinion. However, Camzyos has completely changed this situation. Two large-scale, phase 3 clinical trials conducted as randomized controlled trial (RCT) have confirmed the significant effect of Camzyos. Consequently, ESC guidelines recommend Camzyos with the highest evidence level A for the first time in treatment options. American College of Cardiology (ACC) and the American Heart Association (AHA) are preparing to update their guidelines. Furthermore, based on this phase 3 trial evidence, the U.S. FDA granted Camzyos Breakthrough Therapy Designation (BTD) and approval. Meanwhile, the efficacy of Camzyos was demonstrated through Phase 3 EXPLORER-HCM trials. In this trial, Camzyos improved primary endpoints, which were the patient’s symptoms (NYHA classification) and exercise capacity measured with peak oxygen uptake (pVO2), more than twofold compared to the placebo. 20% of the patients treated with Caymzyos met the NYHA classification and pVO2 improvements. It also reduced the LVOT outflow obstruction index by four-fold after exercise. 7 out of 10 patients who received Camzyos treatment had improved indexes and ended up not considering surgery, and they maintained the effects for 30 weeks.