- LOGIN
- MemberShip
- 2026-07-30 22:46:33
- How will the new drug review process change with fee hike?
- by Lee, Hye-Kyung | translator Alice Kang | 2024-11-06 05:52:15
The Ministry of Food and Drug Safety (MFDS) announced plans to raise the fee for new drug approvals to KRW 410 million from January 1 next year.
The agency has prepared the approval and review process to implement the plan and has begun collecting industry opinion.
According to MFDS’s press corp coverage, the MFDS recently delivered a revised version of the “Operating Procedures for Approval and Examination of New Drug Products (Guideline for public officials)” to the pharmaceutical and biotech industries and relevant associations.
The guidelines contain specific measures to implement the “Innovative Measures for New Drug Approval” that was announced by the MFDS on Sept.
9, including the operation of a dedicated review team for each product and prioritizing GMP-GCP on-site inspections.
“Upon the announcement of the rise in the approval fee of new drugs under the Innovative Plan for New Drug Approval, the pharmaceutical industry raised the question of whether the actual approval period will be shortened,” said an MFDS official.
”By recruiting reviewers, GMP inspections will be prioritized to be completed within 90 days from the date of the application receipt, and pre-registration of supplementary materials and explanatory meetings will reduce unnecessary delays in the approval process, dramatically shortening the approval period for new drugs from 420 days to 295 days.”
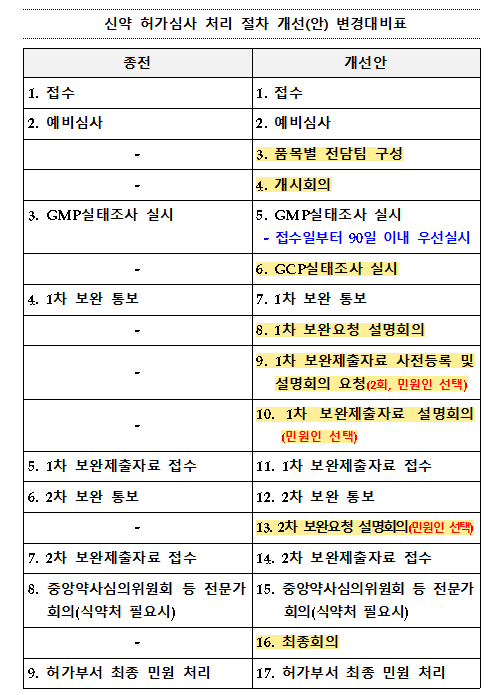
From next year, when a new drug approval application is filed, MFDS will conduct an independent preliminary review within 7 days.
During the preliminary review period, 80% of the fee will be refundable, and the MFDS will check the submission requirements.
If there are any missing materials during this process, the applicant will have 14 days to submit them before an “initiation meeting” is held.
Given the overall review timeline, the preliminary review process is crucial as data not submitted by the initiation meeting will be difficult to submit later and the application will be categorized as an application that requires primary supplementary data.
A key part of this process is the establishment of a dedicated team for each item before the initiation meeting.
The head of the approval division will lead the team, and a representative from the approval division will take charge as each item’s manager, and the team will be composed of representatives from ▲safety, efficacy, ▲quality control, ▲GMP (good manufacturing practice), and ▲GCP (good clinical practice).
In addition, an initiation meeting is held within 14 days of receipt to coordinate the entire approval and review process and to provide the applicant with the necessary materials and procedures.
The initiation meeting may be held face-to-face, via video, or in parallel, which is expected to increase the efficiency of the new drug review process.
According to the proposed Operating Procedures for Approval and Examination of New Drug Products, GMP inspections will be conducted within 90 days from the date of application receipt, and GCP inspections will be conducted within 60 days after the first supplement.
By accelerating the inspection schedule compared to the existing procedure, the verification required for the manufacturing and clinical management procedure will be completed at an earlier stage.
This is intended to speed up the approval process and ensure adequate quality control.
A number of improvements have also been made to the supplement requests.
Previously, there were only first and second supplementary notifications, but the reform introduces a first supplementary request briefing session and a second supplementary request briefing session to ensure that the applicant fully understands the supplementary request.
In particular, the explanatory meetings provide detailed information on the scope and requirements of the required materials and provide time to answer the applicant’s questions.
In particular, for the first round of supplemental data submissions, a pre-registration process and a request for an information session can be made by the applicant.
Applicants can pre-register their supplementary data and request the KFDA to hold an explanatory meeting.
This allows the adequacy of the supplementary data to be reviewed in advance, reducing additional supplementary work and increasing the efficiency of the approval process.
In addition, a final meeting will be held 5 days before the final approval target date to discuss the results of the approval review.
“In addition to the effect of shortening the approval period, the revised procedure will improve the transparency of the approval and review process and help pharmaceutical and biotech companies develop new drugs efficiently while protecting the public's health,” said an MFDS official.
”We will collect opinions from relevant associations and industries to prepare the final draft and continue to optimize and implement the approval review service for the development of the domestic pharmaceutical industry.”
-

- 0
댓글 운영방식은
댓글은 실명게재와 익명게재 방식이 있으며, 실명은 이름과 아이디가 노출됩니다. 익명은 필명으로 등록 가능하며, 대댓글은 익명으로 등록 가능합니다.
댓글 노출방식은
댓글 명예자문위원(팜-코니언-필기모양 아이콘)으로 위촉된 데일리팜 회원의 댓글은 ‘게시판형 보기’와 ’펼쳐보기형’ 리스트에서 항상 최상단에 노출됩니다. 새로운 댓글을 올리는 일반회원은 ‘게시판형’과 ‘펼쳐보기형’ 모두 팜코니언 회원이 쓴 댓글의 하단에 실시간 노출됩니다.
댓글의 삭제 기준은
다음의 경우 사전 통보없이 삭제하고 아이디 이용정지 또는 영구 가입제한이 될 수도 있습니다.
-
저작권·인격권 등 타인의 권리를 침해하는 경우
상용 프로그램의 등록과 게재, 배포를 안내하는 게시물
타인 또는 제3자의 저작권 및 기타 권리를 침해한 내용을 담은 게시물
-
근거 없는 비방·명예를 훼손하는 게시물
특정 이용자 및 개인에 대한 인신 공격적인 내용의 글 및 직접적인 욕설이 사용된 경우
특정 지역 및 종교간의 감정대립을 조장하는 내용
사실 확인이 안된 소문을 유포 시키는 경우
욕설과 비어, 속어를 담은 내용
정당법 및 공직선거법, 관계 법령에 저촉되는 경우(선관위 요청 시 즉시 삭제)
특정 지역이나 단체를 비하하는 경우
특정인의 명예를 훼손하여 해당인이 삭제를 요청하는 경우
특정인의 개인정보(주민등록번호, 전화, 상세주소 등)를 무단으로 게시하는 경우
타인의 ID 혹은 닉네임을 도용하는 경우
-
게시판 특성상 제한되는 내용
서비스 주제와 맞지 않는 내용의 글을 게재한 경우
동일 내용의 연속 게재 및 여러 기사에 중복 게재한 경우
부분적으로 변경하여 반복 게재하는 경우도 포함
제목과 관련 없는 내용의 게시물, 제목과 본문이 무관한 경우
돈벌기 및 직·간접 상업적 목적의 내용이 포함된 게시물
게시물 읽기 유도 등을 위해 내용과 무관한 제목을 사용한 경우
-
수사기관 등의 공식적인 요청이 있는 경우
-
기타사항
각 서비스의 필요성에 따라 미리 공지한 경우
기타 법률에 저촉되는 정보 게재를 목적으로 할 경우
기타 원만한 운영을 위해 운영자가 필요하다고 판단되는 내용
-
사실 관계 확인 후 삭제
저작권자로부터 허락받지 않은 내용을 무단 게재, 복제, 배포하는 경우
타인의 초상권을 침해하거나 개인정보를 유출하는 경우
당사에 제공한 이용자의 정보가 허위인 경우 (타인의 ID, 비밀번호 도용 등)
※이상의 내용중 일부 사항에 적용될 경우 이용약관 및 관련 법률에 의해 제재를 받으실 수도 있으며, 민·형사상 처벌을 받을 수도 있습니다.
※위에 명시되지 않은 내용이더라도 불법적인 내용으로 판단되거나 데일리팜 서비스에 바람직하지 않다고 판단되는 경우는 선 조치 이후 본 관리 기준을 수정 공시하겠습니다.
※기타 문의 사항은 데일리팜 운영자에게 연락주십시오. 메일 주소는 dailypharm@dailypharm.com입니다.
- “Drug pricing intervention contributes to the crisis in essential healthcare and shortage of drugs”
- Reporter's view | Lee, Jeong-Hwan






