- LOGIN
- MemberShip
- 2026-05-01 23:17:58
- Opinion
- [Reporter’s View] Korean drugs go international this year
- by Son, Hyung Min Jan 14, 2025 05:56am
- Last year, new drugs developed by domestic pharmaceutical companies recorded remarkable achievements overseas. Hanmi Pharmaceutical's Rolontis (U.S. brand name Rolvedon) has continued to grow in the U.S. market. Rolontis is a neutropenia treatment developed by Hanmi Pharmaceutical and became the 33rd new domestic drug to be approved in Korea in March 2021. The drug was also approved in the U.S. in September of the same year. Since its launch in the U.S. in the fourth quarter of 2022, Rolontis has posted cumulative sales of USD 110.3 million (approximately KRW 155 billion). SK Biopharm's epilepsy drug Xcopri has also shown success in the U.S. market. Officially launched in the U.S. market in May 2020, Xcopri generated sales of KRW 78.2 billion in 2021, KRW 169.2 billion in 2022, and 270.8 billion in 2023. SK Biopharm expects Xcopri’s sales to have exceeded the KRW 400 billion mark last year. This year, Yuhan Corp’s new cancer drug Leclaza will be launched in the U.S. and European markets. Leclaza was approved in the U.S. and Europe last year for the treatment of non-small cell lung cancer in combination with Janssen's targeted therapy Rybrevant. The combination is currently recommended in the National Comprehensive Cancer Network (NCCN) guidelines. Leclaza is expected will be able to break the dark history of domestic anti-cancer drugs. In the past, SK Chemicals' Sunpla, the first new domestic drug, Hanmi Pharmaceutical's Olita, and Samsung Pharm’s Riavax have challenged the market dominated by foreign anticancer drugs, but they have left the market through cancelation or voluntary withdrawal. In contrast, Leclaza has gone from strength to strength, receiving domestic approval, and then global approval. HLB’s rivoceranib also seeking approval from global regulators this year. In May of last year, the company and its Chinese partner Jiangsu Hengrui Medicine received a complete response letter (CRL) from the FDA, but a was found to have no deficiencies in the recent review. The combination of rivoceranib and Jiangsu Hengrui Medicine’s immuno-oncology drug camrelizumab has shown the longest survival benefit among first-line liver cancer treatments. Domestic new drugs that emerged last year are also eyeing the global market. Onconic Therapeutics is in the process of licensing out its technology for its P-CAB-based gastroesophageal reflux disease drug Jaqbo. Vivozon Pharmaceutical's non-narcotic painkiller Unafra is also emerging as an alternative to narcotic painkillers. The domestic companies need to keep in mind not only the domestic market but also global expansions for survival. Last year, the global pharmaceutical industry market was valued at USD 1.44 trillion (about KRW 1901.6 trillion). Compared to Korea's market size of USD 18.2 billion (about KRW 25.5 trillion), the global market is about 79 times larger. To put it bluntly, none of the homegrown new drugs developed to date can be called a “global blockbuster drug.” Global blockbuster drugs usually refer to products that post annual sales of more than KRW 1 trillion, but a sufficient market is required to achieve such sales. A small market cannot generate KRW 1 trillion in sales. In the end, going global is not an option but a necessity to grow further in the domestic pharmaceutical bio-industry. It is necessary to consider various research institutions such as clinical sites with the global market in mind from the initial stages of development. If homegrown new drugs successfully go global, this is expected to drive the performance of later drugs. In addition, if the domestic pharmaceutical and biotech industry shows results overseas, a virtuous cycle of reinvestment in R&D and investment in promising biotech companies is expected to be established. Currently, various domestic new drugs such as K-CAB and Fexuclue are conducting clinical studies to enter the global market. If domestic pharmaceutical companies' new drugs pave the way this way, it will be easier for latecomers to enter the market. I hope this year domestic drugs will show performance not only on the domestic stage but also on the international stage.
- Company
- 'Leclaza·Rybrevant comb therapy' wins nod in KOR
- by Son, Hyung Min Jan 14, 2025 05:56am
- YuhanThe approval of Leclaza plus Rybrevant combination therapy in South Korea has expanded treatment options for patients with non-small cell lung cancer (NSCLC). As Leclaza combination therapy has shown a positive overall survival (OS) result, it is highly likely to be the first-line standard therapy for treating EGFR-positive NSCLC. According to industry resources on January 13, the Ministry of Food and Drug Safety (MFDS) granted approval for Leclaza plus Rybrevant combination therapy as the first-line treatment for EGFR-positive NSCLC. Following the approval, Leclaza plus Rybrevant combination therapy can be used in adult patients with advanced or metastatic NSCLC who have EGFR exon 19 deletion mutation or exon 21 (L858R) point mutation. Following the approvals in the United States and Europe last year, Leclaza plus Rybrevant combination therapy was approved in South Korea as the first in Asia. The US Johnson & Johnson aims to receive approval for Leclaza plus Rybrevant combination therapy in Japan and China. The basis of approval is the MARIPOSA Phase 3 trial. The study involved 1,074 patients with locally advanced or metastatic NSCLC. The median age of patients was 63, and over half of the participants were Asian (59%). Brain metastases occurred in 41% of the participants. Patients were randomly assigned in a 2:2:1 ratio to Leclaza plus Rybrevant combination therapy, Tagrisso monotherapy, and Leclaza monotherapy. The primary endpoint was progression-free survival (PFS), a period without disease progression, and the secondary primary endpoints were overall survival (OS), PFS after the first follow-up treatment (PFS2), and an overall response rate (ORR). The clinical trial results showed the Leclaza plus Rybrevant combination therapy group had 23.7 months of PFS. The Lecalza monotherapy group had 18.5 months of PFS, which was longer than the 16.6 months of the Tagrisso monotherapy group. The Leclaza plus Rybrevant combination therapy group had 30% lower disease progression and death risk compared to the Tagrisso monotherapy group. In the MARIPOSA study, adverse reactions of over Grade 3 were reported as 75% and 43% in the Leclaza plus Rybrevant combination therapy group and the Tagrisso monotherapy group, respectively. Significant adverse reactions were reported at 49% and 33%. Treatment-related adverse reactions in the Leclaza plus Rybrevant combination therapy group mainly were Grade 1-2, such as nail infection and rash. Recently presented OS results showed that Leclaza plus Rybrevant combination therapy is superior to the Tagrisso monotherapy. Johnson & Johnson explained that Leclaza plus Rybrevant combination therapy had statistically significant extended median OS over 1 year compared to Tagrisso therapy. Since Tagrisso recorded an OS of 38.6 months in the FLAURA study, the basis of approval, the OS of Leclaza plus Rybrevant combination therapy will likely be around 50 months. The current results indicate an improvement over previous clinical data. Previously, the Leclaza plus Rybrevant combination therapy has demonstrated the efficacy regarding the primary endpoint, PFS. The secondary endpoint, OS, had shown a favorable trend compared to Tagrisso. If the efficacy of Leclaza plus Rybrevant combination therapy is confirmed in OS, in addition to PFS, then it will become the next standard therapy for EGFR-positive NSCLC.

- Company
- Samsung Bioepis partners with Teva for Soliris biosimilar
- by Cha, Jihyun Jan 14, 2025 05:56am
- Pic of Samsung Bioepis Headquarters Samsung Bioepis announced on the 12th that it has signed a commercialization partnership agreement with the multinational pharmaceutical company Teva Pharmaceutical Industries to launch Epysqli, a biosimilar of the rare disease treatment Soliris (eculizumab), in the U.S. market. Epysqli is the first hematology biosimilar developed by Samsung Bioepis. Its original drug, Soliris was developed by Alexion, a U.S. company specializing in rare disease treatments. It is used for rare intractable diseases such as paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS). It is an ultra-high-priced drug that posted global sales of approximately KRW 5 trillion in 2023. Samsung Bioepis obtained the U.S. Food and Drug Administration (FDA) approval for Epysqli for PNH and aHUS in July last year. In November last year, the company expanded the indication to include generalized myasthenia gravis (gM) through additional FDA approval. Under the agreement, Samsung Bioepis will launch Epysqli in the U.S. market through Teva, and the release is expected in the first half of this year. Samsung Bioepis will be responsible for the production and supply of Epysqli. Teva will be responsible for marketing and sales activities in the U.S. “Epysqli is a drug that best realizes the essence of biosimilar development as it enables expanded access to ultra-high-priced biologics,” said Kyung-Ah Kim, CEO of Samsung Bioepis. “We look forward to working closely with Teva to improve the lives of patients with rare diseases in the U.S.” “We are pleased to partner with Samsung Bioepis to help expand patient access,” said Chris Fox, Executive Vice President US Commercial at Teva Pharmaceuticals. ”We look forward to leveraging our commercial capabilities with Samsung Bioepis to expand treatment access for our patients.”

- Company
- Will biotech companies continue 'K-Bio' success?
- by Moon, sung-ho Jan 13, 2025 05:53am
- Korean pharmaceutical and biotech companies have successfully outlicensed new drugs in various fields, including cancer, autoimmune disease, and Alzheimer's disease. However, compared to the year before, analysis suggests that the number of out-licensed cases decreased. Despite such a slowdown, companies focusing on global new drug development trends, including Alzheimer's disease treatment and antibody-drug conjugates (ADC), have drawn interest from the industry following achieving grand-size contracts. We analyzed major out-licensing cases from Korean pharmaceutical and biotech companies in 2024 and explored the potential of future out-licensing possibilities for 2025. According to pharmaceutical and biotech companies on December 30, 2024, the number of out-licensing transactions in the Korean pharmaceutical and biotech industry for 2024 was 14. Compared to last year's 18 cases, it is a slight decrease. However, analyzing out-licensed transactions, companies that have achieved this by focusing on global new drug development trends stand out. First, LigaChem Biosciences and Orum Therapeutics have successfully out-licensed for two consecutive years, and becoming blu-chip companies in the pharmaceutical and biotech industry. In October 2024, LigaChem Biosciences and a Japanese pharmaceutical company, Ono-Pharma, signed two technology transfer agreements, including the L1CAM-targeting ADC candidate 'LCB97.' According to the contract between both companies, the specific upfront payment remains undisclosed. Two agreements total over US$700 million (KRW 943.5 billion). L1CAM, targeted by LCB97, is a protein expressed in several solid cancers, including lung cancer, pancreatic cancer, and colorectal cancer. LigaChem Biosciences' proprietary ConjuAll linker was used in the development of LCB97. ADC is made of a linker, a payload, and an antibody. The ConjuAll linker is known to overcome the issue of releasing cytotoxic drugs into the blood and attacking healthy cells. In addition to the LCB97 deal, LigaChem Biosciences and Ono-Pharma signed an agreement to transfer the ADC platform technology designed to target dual targets. Based on the contract, using LigaChem Biosciences' platform technology, Ono-Pharma secured rights to discover·develop ADC candidates for multiple targets. Orum Therapeutics has proven its R&D capacity by successfully out-licensing the DAC platform to global pharmaceutical companies. In July last year, the company signed an out-licensing agreement with the U.S.-based biotechnology company Vertex Pharmaceuticals for its DAC. Orum Therapeutics received a US$15 million upfront payment and will receive up to $310 million in potential option fees, milestone payments per target, and tiered royalties. Notably, the company has succeeded in advancing the development of new drugs for cancer, autoimmune diseases, and Alzheimer's disease. HK inno. N signed a license agreement with Navigator Medicines, a US-based pharmaceutical company, for 'IMB-101,' a novel drug candidate for the treatment of autoimmune diseases. The contract totaled US$940 million (approximately KRW 1.3 trillion), including an up-front payment of US$20 million (KRW 27.6 billion). Navigator Medicines secured global development and sales rights through this agreement, excluding Asia. AprilBio also successfully out-licensed 'APB-R3,' an autoimmune disease candidate, to a US-based new drug developer, Evommune. It has been contracted for up to US$475 million (approximately KRW 655 billion), including a non-refundable upfront fee of US$15 million (KRW 20.7 billion), with a separate royalty payment for sales. Aribio successfully out-licensed 'AR1001,' a new drug candidate for Alzheimer's disease, to a Chinese pharmaceutical company in March 2024. Aribio received an upfront payment of KRW120 billion. The company made an advancement in developing an oral new drug candidate for Alzheimer's disease amid increased interest in Alzheimer's disease treatment following the release of Leqembi (lecanemab). AR1001 targets the underlying causes of Alzheimer’s disease through multimodal mechanisms, such as PDE5 and toxic proteins. This new drug candidate is based on Mvix (mirodenafil), which is similar to Viagra and a treatment for erectile dysfunction. Jai Jun Choung, CEO of Aribio, said, "The global market is closely watching Aribio's AR1001." Choung added, "Since Aribio is the first Korean biotech company to conduct a global phase 3 trial in the field, we are striving to achieve success." Particularly this year, Korean companies have not been limiting their goals in exporting new drug candidates. For instance, 'Alteogen' has achieved multiple successes in out-licensing its technology to change the type of anticancer agent formulation. In November 2024, Alteogen signed an exclusive license agreement with Daiichi Sankyo to develop and sell the new ADC drug 'Enhertu' of a subcutaneous (SC) formulation. The company received a non-refundable upfront payment of US$20 million. Enhertu, an antibody-drug conjugate (ADC) jointly developed by Daiichi Sankyo and AstraZeneca, has been approved for the treatment of HER2-positive breast cancer and gastric cancer. As Enhertu demonstrated to be effective in HER2 mutant NSCLC and HER2-low breast cancer, its indication was expanded. Soon Jae Park, CEO of Alteogen, said, "We will be able to provide an alternative administration route by developing a subcutaneous formulation of Enhertu by signing a partnership with Daiichi Sankyo and using ALT-B4," Park added, "We hope to provide a wide variety of treatment options to patients by utilizing ALT-B4 to numerous treatments in the future." Global pharmaceutical companies with major immunotherapy strive to change conventional injectables to subcutaneous formulations. In this process, Alteogen is co-developing the SC formulation of Keytruda with MSD. It has been reported that Alteogen achieved growth through pursuing the Enhertu formulation change project. Conventional anti-cancer treatments are primarily intravenous (IV) therapy, and the administration takes more than one hour. Anti-cancer treatments of SC formulation are expected to improve patient convenience since they can significantly reduce the administration duration to within 10 minutes. Professor Byoung Chul Cho (Director of the Lung Cancer Center at Yonsei Cancer Hospital) said, "The United States provides incentives to using injections, and the amount of incentives is the same between IV injectable or SC injectable," and explained, "There is no need to maintain IV formulation injectables, which commonly induce injection-associated adverse reactions." With these advancements, companies adopting differentiated strategies in global new drug development trends are likely achieve additional out-licensing cases by 2025. Analysis suggests that the global pharmaceutical industry will likely continue pursuing pipelines in various fields, such as autoimmune diseases, radiopharmaceuticals, cell therapies, and Alzheimer's treatments. If companies, like the case of Alteogen, also pursue a differentiation strategy as an advantage, they are expected to maintain strong competitiveness. Major global pharmaceutical companies, including Vertex Pharmaceuticals, Gilead Sciences, AbbVie, Lilly, Merck, and Sanofi, have actively expanded their pipelines through M&A this year. The largest M&A deal of the year was Vertex Pharmaceuticals's acquisition of Alpine Immune Sciences. A contract worth US$4.9 billion (approximately KRW 7.03 trillion), including milestone achievements, has been signed. Based on the agreement between the two US-based global pharmaceutical companies, Vertex has secured povetacicept. Povetacicept is a bispecific antibody targeting APRIL, which is involved in the proliferation of BAFF for B-cell activation. Vertex also signed an out-licensing agreement with the Korean biotech company Orum Therapeutics. Analysis suggests a high likelihood that global pharmaceutical companies will continue efforts to secure future growth opportunities as patents for their therapies expire, suggesting that Korean pharmaceutical and biotech companies have significant potential for out-licensing. However, investor sentiment has declined in the pharmaceutical and biotech industry this year, posing risks to clinical research efforts and potentially hindering technology export achievements next year. Seung-Kyou Lee, Vice President of the Korea Biotechnology Industry Organization (Korea Bio), said, "In response to the urgent issue of decreased investment in the biotech sector, we strive to actively facilitate domestic and international investor matching and operate a demand-supply company committee." Lee added, "We need measures to identify and support solutions for the business and policy demands of biotech companies working in various fields."
- Company
- Samsung Bioepis to indirectly sell Soliris biosimilar in US
- by Cha, Jihyun Jan 13, 2025 05:53am
- Samsung Bioepis has signed a partnership agreement with the multinational pharmaceutical company Teva Pharmaceuticals to commercialize biosimilars of rare disease treatments in the United States. Samsung Bioepis will launch the biosimilar through Teva in the U.S. market in the first half of this year. Unlike in Europe, where Samsung Bioepis has been selling its drug directly, the company's strategy in the U.S. has been to forge a partnership to enter the market. Analysts say this is due to the complex structure of the U.S. drug market, which is centered on private insurance, and the cost burden of direct sales. Samsung Bioepis enters into a partnership with Teva to commercialize Epysqli... to launch in the U.S. in the first half of the year Samsung Bioepis According to industry sources on the 13th, Samsung Bioepis recently signed a commercialization partnership agreement with Teva to launch Epysqli, a biosimilar version of the rare disease treatment Soliris (eculizumab), in the US market. Soliris was developed by Alexion, a U.S. developer specializing in rare disease therapies. It received marketing authorization from the U.S. Food and Drug Administration (FDA) in March 2007 and the European Commission (EC) in June of the same year. In 2021, AstraZeneca acquired Alexion and took over the rights to Soliris. Soliris is indicated for rare diseases, including paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), neuromyelitis optica syndrome disorder (NMOSD) generalized myasthenia gravis (gMG). In 2023, Soliris reported global sales of USD 3.145 billion (approximately KRW 4 trillion). Of this, the U.S. market is estimated to be worth about KRW 2.3 trillion and the European market is estimated to be worth about KRW 1 trillion. Epysqli is the first hematology biosimilar developed by Samsung Bioepis. Samsung Bioepis obtained the EC marketing authorization for Epysqli for PNH in May 2023 and launched it in the European market in July of the same year. In Korea, it has been on the market since January last year after receiving approval from the Ministry of Food and Drug Safety. In the U.S., the FDA approved Epysqli for PNH and aHUS in July last year. In November last year, the FDA approved an additional indication for its treatment of gMG. Under the agreement, Samsung Bioepis will launch Epysqli in the U.S. market through Teva. The launch is expected in the first half of this year. Samsung Bioepis will be responsible for the production and supply of Epysqli. Teva will be responsible for the drug’s marketing and sales activities in the US. This agreement brings Samsung Bioepis' total number of international partners to four. Samsung Bioepis currently has forged commercialization partnerships with Biogen, Organon, and Sandoz in overseas markets. The partners have exclusive rights to sell the contracted products in the targeted territories and pay Samsung Bioepis milestones and sales commission royalties. Under the new agreement, Samsung Bioepis will receive milestone payments from Teva. Both companies will also share a percentage of product sales revenue. Specific milestone amounts and revenue-sharing percentages for each company were not disclosed. Decided to forge partnership in the U.S., unlike selecting direct sales in Europe, considering the U.S drug structure and costs Epysqli is the only product that contains the company’s name in the product name and is also Samsung Bioepis’ first direct sales product. Epysqli indications, PNH, and aHUS are among those ultra-rare diseases that have a very small number of patients. Samsung Bioepis decided to establish a direct sales system for the first time for Epysqli among all its products, believing that it could fully carry out sales activities with its small number of salespeople. By selling directly without distributing through partners, Samsung Bioepis can reduce commission expenses and increase profitability. Domestic companies pay around 30% to 40% of sales on average as commission to partners when expanding overseas. Another advantage is that it gives the company more market control. In Europe, the effects of the company’s direct sales are slowly showing results. As of the third quarter of last year, Epysqli held the No. 1 market share for eculizumab-based biosimilars in Germany and Italy. It has also secured contracts with the largest procurement group in France (UniHA) and the Dutch government. Samsung Bioepis Nevertheless, Samsung Bioepis' decision not to directly market Epysqli in the U.S. was based on the complex drug structure in the U.S. and the initial direct marketing costs. The European drug market uses a tender system, making it relatively easy to enter. On the other hand, the U.S. drug market has a complex structure centered on private insurance, so it would have been difficult for the company to go direct. The cost of establishing an initial direct sales system may also have been a burden. While the profitability of a direct sales system increases with the number of products sold, it requires large fixed costs to establish a local subsidiary and hire specialized sales and marketing personnel. Will seek to accelerate its entry into the U.S. market with its lower drug price than the original (KRW 400 million/year) and product competitiveness Samsung Bioepis aims to speed up its expansion into the U.S. market by capitalizing on Epysqli’s price competitiveness and product competitiveness. Soliris is an ultra-high-priced drug that costs about KRW 400 million per year. Epysqli’s domestic drug price is set at KRW 2.51 million per vial. This is half the price of the original drug, which costs KRW 5.13 million. This is also about 30% lower than the price of Soliris, which was newly introduced in April last year at KRW 3.6 million. The idea is to gain an advantage over the original through its price competitiveness and expand patient access. Being ‘sorbitol-free' is also considered a competitive advantage of Epysqli. Sorbitol is a substance that helps improve the stability of medicines. However, it can cause reactions in patients with fructose intolerance, who cannot digest fructose precursors such as fructose or sugar. In Europe, medications containing sorbitol are banned for people with fructose intolerance. The U.S. has no such prohibition. However, industry experts believe that given the potential for reactions in some patient populations, it is likely that clinicians will favor products without sorbitol. Epysqli’s competitor, Amgen's Soliris biosimilar Bkemv contains sorbitol. However, the original company’s patent defense strategy may serve as a variable. Alexion is marketing Ultomiris, a once-eight-weekly dosing version of Soliris that offers improved dosing convenience. Both Soliris and its biosimilar are administered intravenously every 2 weeks. In this regard, Samsung Bioepis believes that there is enough market potential as there is still a demand for Soliris in the medical field and there are many markets where Ultomiris has not entered yet. “Teva is a generic and biosimilar company with extensive sales and marketing infrastructure in the U.S. market,” said a Samsung Bioepis official. ”We chose Teva as our commercialization partner for Epysqli in the U.S. market because of its experience and expertise in the U.S. market.” The official added, “The market for Soliris is much larger in the U.S. than in Europe, and we are excited to see how Epysqli will sell in the U.S. given its success in Europe.”
- Company
- Wegovy can be prescribed in tertiary hospitals in KOR
- by Eo, Yun-Ho Jan 13, 2025 05:53am
- The obesity drug ‘Wegovy’ can now be prescribed in tertiary hospitals in Korea. According to industry sources, Novo Nordisk Korea's Wegovy (semaglutide) has passed the drug committees (DCs) of Korrea’s “Big 5 medical institutions,” including Samsung Medical Center, Seoul National University Hospital, and Sinchon Severance Hospital. Wegovy was approved by the U.S. Food and Drug Administration (FDA) in June 2021 and was granted marketing authorization in Korea in October last year. The once-weekly obesity treatment is approved as an adjunct to a reduced-calorie diet and increased physical activity for weight management, including weight loss and weight maintenance, in adult patients. Specifically, it is indicated for use in obese patients with an initial body mass index (BMI) of 30 kg/m2 or greater or in overweight patients with one or more weight-related comorbidities and a BMI of 27 kg/m2 or greater but less than 30 kg/m2. Obesity is associated with chronic diseases such as hypertension and type 2 diabetes and is known to contribute to the development of cardiovascular disease and cancer. The number of obese people in Korea is steadily increasing, with the obesity rate in Korea reaching 37.1% in 2021. The starting dose of Wegovy is 0.25 mg once a week and is gradually increased to a maintenance dose of 2.4 mg once a week for 16 weeks. Wegovy is available in five dosage forms: 0.25 mg, 0.5 mg, 1.0 mg, 1.7 mg, and 2.4 mg. The efficacy and safety profile of Wegovy was demonstrated through the large-scale STEP trial. In the multinational trial that involved 1,961 adult overweight or obese patients, the Wegovy-treated group demonstrated a mean weight loss of 14.9% from baseline over 68 weeks (1,306 patients) compared to 2.4% in the placebo group (655 patients), demonstrating a significant difference. All overweight or obese patients in the trial were on a reduced-calorie diet and increased physical activity.
- Policy
- Will 'Lorviqua' secure a deal on drug price negotiations?
- by Lee, Tak-Sun Jan 13, 2025 05:53am
- Product photo of LorviquaThe reimbursement expansion case for Lorviqua (lorlatinib, Pfizer), which was stalled during the drug price negotiation last year, has passed the Drug Reimbursement Evaluation Committee (DREC) of the Health Insurance Review and Assessment Service (HIRA) under a conditional term. It draws attention to whether it will succeed this round. Pfizer wants the termination of the Risk Sharing Agreement (RSA) and the reimbursement expansion for this drug, so the focus is on whether the company will reach an agreement with the insurance authority. The DREC meeting on January 9 ruled that Lorviqua would be appropriate for expanded reimbursement scope if the company accepted the drug price below the evaluated amount. Consequently, if Pfizer agrees with the drug price below the evaluated amount, it will proceed with the negotiation phase with the NHIS. Lorviqua is under consideration for reimbursement expansion as a first-line treatment for patients with anaplastic Lymphoma Kinase (ALK)-positive and metastatic non-small cell lung cancer. The drug passed the DREC last year, but it failed after negotiating with the National Health Insurance Service (NHIS). It has been reported that the company differed in opinion regarding adjusting the cap for reimbursement ceiling type during the negotiation process. Pfizer wanted to terminate the RSA and switch to general medicine reimbursement. When it was initially approved for reimbursement, Lorviqua was categorized as a reimbursement ceiling type under the cost-effectiveness evaluation waiver system. However, it was approved for being cost-effective after receiving the economic evaluation by the HIRA. However, the company failed to reach an agreement at the drug pricing negotiation because RSA was not terminated. Following the announcement of failed negotiations, doctors and patients called for immediate reimbursement listings. During the parliamentary audit held in October, Rep. Han Ji-ah, a member of the People Power Party, stressed the importance of reimbursement expansion while introducing a male patient's case. Regarding this, Lee Joongkyu, Director of the National Health Policy at the Ministry of Health and Welfare (MOHW), said, "We will help patients receive benefits by quickly reaching an agreement as soon as possible." The NHIS recently revised the RSA guidelines regarding the termination clause. Cost-effectiveness-evaluated drugs like Lorviqua that have demonstrated cost-effectiveness through the DREC review, companies and NHIS can renegotiate to adjust reimbursement ceilings, reset expected claim amounts, or modify·terminate total expenditure-capped RSAs. This opens the possibility of terminating total expenditure-capped RSAs through the renewal of the agreement. Drugs categorized as a basic refund type RSA can be terminated early when a pharmaceutical company wishes to terminate. In the case of Lorviqua, the company applied for RSA termination before the revision, early termination may be possible, even if it is not for renewing contract negotiations. Prior to the revision, drugs were not categorized into specific types that could be terminated early. Consequently, when the company begins negotiation with the NHIS, there will be a discussion about RSA termination and reimbursement expansion. The remaining issue is whether both parties could agree on the drug price without financial burden while allowing RSA termination. In the CROWN trial, Lorviqua reduced the disease progression and death risk by 81% compared to crizotinib, and 60% of treated patients were alive without disease progression after five years. The results indicate that reimbursement of the drug is necessary. The industry draws attention to whether the discussion for expanding Lorviqua's reimbursement, which previously failed in drug pricing negotiations, will lead to an agreement this year.
- Policy
- New criteria set for unstable supply and demand drugs
- by Lee, Tak-Sun Jan 13, 2025 05:53am
- The Health Insurance Review and Assessment Service has decided to clarify the criteria for evaluating adjustment applications for drugs with unstable supply and demand and strengthen preliminary management of high-priced anticancer drugs. HIRA announced so on the 9th while issuing a prenotification of the “Partial Amendments to the Evaluation Criteria and Procedures for Whether a Drug is Eligible for Medical Benefits” and the “Partial Amendments to the Operating Regulations of the Severe Disease Deliberation Committee.” The deadline for submitting opinions on the criteria is the 15th. First, in the prenotification of the “Partial Amendments to the Evaluation Criteria and Procedures for Whether a Drug is Eligible for Medical Benefits,” drugs with unstable supply and demand were newly added as evaluation criteria for adjustment applications. “In order to support the stabilization of drug supply in the context of health security, HIRA has established criteria for the evaluation of adjustment applications to be eligible for negotiations in cases where there is a request for cooperation from central administrative agencies regarding the supply of drugs due to an infectious disease crisis or an urgent shortage of supply, and in cases where an increase in drug price is necessary due to diversification of raw material supply as a national essential medicine.” Accordingly, the following items will be added to the evaluation criteria for adjustment applications: ▲ if there is a request for cooperation from central administrative agencies related to the supply of drugs due to an infectious disease crisis or urgent shortage of supply ▲ if a national essential medicine has diversified its supply of raw materials. The amendment to the regulation is a follow-up to the government's “Plan to Reflect the Innovative Value of New Drugs and Improve the Drug Price System for Health Security.” In particular, the evaluation criteria for adjustment applications to grant drug pricing premiums for national essential medicines that use domestically produced raw materials seems to have been established. The price of drugs with unstable supply and demand is already being raised through adjustment applications upon the council's recommendation. The amendment to the Operating Regulations of the Serious (Cancer) Disease Deliberation Committee also added a provision for the Head of the department in charge of drug post-management to attend the CDDC meeting. On this, HIRA explained, “The recent increase in the number of new drugs that are expensive and have great uncertainty in terms of efficacy has increased the importance of performance management after reimbursement and expansion. Therefore, the amendment was made to strengthen the practicality of drug management by allowing the head of the department that collects and evaluates the actual effectiveness of drugs to participate in the deliberation stage.” HIRA has recently been promoting the introduction of post-management of high-priced drugs through RWD. It has also established a drug performance evaluation division under the Health Insurance Review and Assessment Policy Research Institute. Prior to the full-scale implementation of the system, it is understood that the amendment was made to preemptively prepare a post-management plan by having the heads of relevant departments participate in the cancer review, the first gateway to the review of anticancer drugs.
- Company
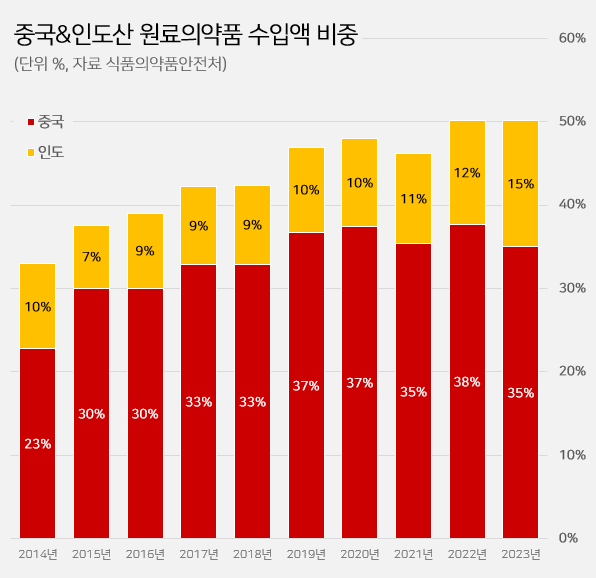
- Imported API from China·India reaches 50%
- by Kim, Jin-Gu Jan 13, 2025 05:53am
- It has been reported that half of the active ingredients (API) imported to South Korea are produced in China and India. The percentage of imported active ingredients originating from China·India surpassed 50% in two consecutive years. The percentage of imported API from India has robustly increased recently. Indian API imports were below 10% in 2018, now expanded to 15.2% in five years. In contrast, the percentage of Chinese API imports slightly dropped from 36.7% to 35.0%. According to the 'Annual report of food and drug statistics for 2024' by the Ministry of Food and Drug Safety (MFDS) on January 11, domestically imported API in 2023 totaled US$ 2.199 billion. Among these, API imports from China·India totaled US$ 1.1 billion. Imports from China·India account for nearly 50.2% of the total imported API. It indicates that half of domestically imported API is from China·India. Analysis suggests that reliance on API produced in China·India has consistently increased. The percentage of imported API from China·India surpassed 40% in 2017, rising to over 50% by 2022. It recorded over 50% in two consecutive years until 2023. The percentage of imported API from China·India: API imports from China and India surpassed 40% in 2017, rising to over 50% by 2022. It recorded over 50% in two consecutive years until 2023. (unit: %, source: Ministry of Food and Drug Saftey). API imports from China have declined, while imports from India have risen. Imported API from China in 2023 totaled US$ 769.76 million, a drop of 16.0% from US$ 916.87 million in 2022. Imported API from India rose by 10.1% from US$ 303.30 million in 2022 to US$ 334.00 million in 2023. Looking into the years before, imported API from India has shown an increasing trend for the past few years. In 2018, imported API from India was US$ 195.56 million, an increase of 70.8% in five years. The percentage of imported API from India was merely 9.5% in 2018, while it rose by 5.7% in five years to 15.2% in 2023. Analysis suggests that as the decrease in imports from China has been compensated by API from India, South Korea has increasingly depended on APIs produced from both countries. As of 2023, besides China·India, there are no imported API from other countries that account for more than 10%. Japan ranks third in the amount of imported API in 2023, totaling US$ 198.85 million (9.0%). Japan's share had sustained over 10% until 2022. Followed by France US$ 170.66 million (7.8%), Italy US$ 123.59 million (5.6%), Germany US$ 120.81 million (5.5%), the U.S. US$ 66.14 million (3.0%), Spain US$ 44.67 million (2.0%), Switzerland US$ 36.77 million (1.7%), and Belgium US$ 31.78 million (1.4%).
- Company
- Sanofi’s Hexaxim is included in NIP from this year
- by Whang, byung-woo Jan 10, 2025 05:52am
- Pic of Hexaxim Sanofi announced on the 9th that Hexaxim, its hexavalent combination vaccine for infants, has been included in Korea’s National Immunization Program (NIP). With its introduction to the NIP, Hexaxim can now be administered free of charge at designated medical institutions under the National Immunization Program for Children. As the first and only hexavalent combination vaccine in Korea (as of January 2025), Hexaxim can simultaneously protect against 6 infectious diseases including hepatitis b in addition to the 5 infectious diseases (diphtheria, tetanus, pertussis, polio, Haemophilus influenzae type b ) that can be prevented by existing pentavalent combination vaccines. Infants who received hepatitis B monovalent vaccines at birth are eligible for the vaccine, which is given in a series of 3 doses at 2, 4, and 6 months of age. However, infants born to hepatitis B-positive mothers will be vaccinated with the pentavalent combination vaccine and hepatitis B monovalent vaccine as before, as it is necessary to prevent vertical infection of hepatitis B. The designated medical institutions for the National Immunization Program can be found on the NIP KDCA website, and it is necessary to check the types of vaccines that can be administered at the designated medical institution before visiting, as the types of vaccines offered may differ depending on the medical institution. The hexavalent combination vaccine for infants has been already recommended as an essential vaccine in more than 40 countries, including Europe, Canada, and Australia. Hexaxim also reduces the total number of doses from 6 to 4 compared to the current schedule that requires separate doses of the pentavalent combination vaccine and hepatitis B monovalent vaccine, rendering it easier for parents and infants to get vaccinated and improving timely immunization rates by reducing delays and missed doses of recommended vaccines. Change in number of doses before and after introduction of Hexaxim In addition, Hexaxim is a ready-to-use formulation that maximizes convenience and efficiency of administration, requires no reconstitution, and is easy to use. This reduces vaccination preparation time and significantly reduces the risk of reconstitution errors, making the process more efficient for healthcare professionals and enabling them to provide safe and rapid immunization services to parents and infants. “The introduction of Hexaxim into the NIP is a recognition of Hexaxim’s public health value, and we are pleased to be able to contribute to easing the financial burden on parents and increasing the efficiency of the healthcare system,” said Hee-Kyung Park, Head of Sanofi Vaccines Business Unit. “As a long-standing public health partner in Korea, Sanofi has contributed to the prevention of infectious diseases in the country. We will continue to be committed to preventing infectious diseases by creating a seamless vaccination environment and the timely provision of vaccines.”