- LOGIN
- MemberShip
- 2026-04-27 13:31:37

- Company
- Adult vaccination in a super-aged society highlighted
- by Whang, byung-woo Aug 13, 2025 06:07am
- South Korea has entered a super-aged society, with its population aged 65 and over exceeding 20%. The need for a response through adult vaccination is being emphasized. With the number of National Immunization Program (NIP) vaccines for adults limited to just influenza and pneumococcal vaccines, there is a growing opinion that the program needs to be expanded. The British Embassy in Seoul, the British Chamber of Commerce in Korea, and GSK Korea jointly hosted the '2025 Healthy Ageing Korea' forum on August 12 to discuss the necessity of adult vaccination. Professor Kwang-il Kim of the Department of Geriatrics at Seoul National University Bundang Hospital Professor Kwang-il Kim of the Department of Geriatrics at Seoul National University Bundang Hospital gave a presentation on 'Adult Vaccination for an Extended Healthy Lifespan'. Professor Kim emphasized the need for a full-lifecycle vaccination program as a healthcare policy to prevent infections and extend healthy lifespans in an aging society. Professor Kim stated, "While the consumption of medical resources and the social burden continue to increase among the elderly population in Korea, there are not many vaccines included in the National Essential Vaccination Program for adults, many of whom have chronic diseases that make them vulnerable to infectious diseases." In fact, NIP vaccines for adults are limited to two types: influenza and pneumococcal. Other adult vaccines are supported through small-scale vaccination projects using local governments' own budgets, leading to issues of fairness between regions. Professor Kim said, "Vaccination in the elderly can contribute to healthy aging by preventing disease-related complications, thereby reducing the medical burden and mortality rates." He added, "Diverse policy and strategic approaches to vaccination are needed for an aging society and healthy aging." Professor Hankil Lee of Ewha Womans University's College of Pharmacy gave a presentation on 'The Value of Adult Vaccination in Response to a Super-Aged Society', highlighting adult vaccination as a crucial public health policy for an aging population. According to Professor Lee, as the effectiveness of vaccination has been proven in the elderly, major overseas countries are including adult vaccination in their NIPs and supporting it with public funds. The UK offers free shingles vaccination for its elderly population, and Japan has been supporting shingles vaccines with a mixed financial structure since April of this year. Professor Hankil Lee of Ewha Womans UniversityProfessor Lee's opinion is that adult vaccination not only protects individuals from infectious diseases but also contributes to improving public health and reducing the socioeconomic burden at a national level, thereby generating economic effects from various perspectives. Professor Lee said, "In Korea, adult vaccination has not been fully recognized as a public good with sufficient social benefits from the perspective of responding to a super-aged society." He emphasized, "The policy gap in adult vaccination continues to exist, and it must be systematically addressed when establishing a comprehensive national vaccination plan in the future." The forum also revealed the results of a cost-benefit analysis of shingles and RSV (Respiratory Syncytial Virus) vaccines in adults in Korea. The analysis showed that for shingles vaccines in individuals aged 50 and over, the Return on Investment (ROI), or socioeconomic benefit relative to cost, was approximately 1.52. For RSV vaccines in the elderly aged 60 and over, the socioeconomic benefit was 1.65. Professor Lee said, "When the socioeconomic benefit exceeds 1, it is considered that a larger social benefit was generated than the cost incurred. This analysis proves that adult vaccination is a public investment that brings long-term socioeconomic benefits, going beyond just disease prevention." Colin Crooks, British Ambassador to the Republic of Korea, added, "While the elderly population is increasing globally, Korea is aging at a particularly fast pace." He continued, "In this situation, this forum was a meaningful opportunity for academia, the government, and the elderly community to discuss the protection of the health rights of the elderly and the necessity of prevention. It is expected to be an opportunity to raise awareness of the importance of prevention-centered public health across society."

- Company
- Tariff impact inevitable for biodrugs’ export to the US
- by Cha, Jihyun Aug 12, 2025 06:16am
- Exports of domestically produced biopharmaceuticals to the US more than doubled last year compared to the previous year. If the Trump administration's tariffs on pharmaceuticals are implemented, the domestic biopharmaceutical export industry is expected to suffer significant damage. According to the “Key Data on the Domestic Biopharmaceutical Industry 2025” published by the Korea Biomedicine Industry Association (KoBIA, President: Jeong-seok Lee) on the 11th, last year's exports of domestically produced biopharmaceuticals to the United States reached USD 686.7 million (approximately KRW 845.9 billion). This represents a 103% increase from the previous year's USD 300.72 million (approximately KRW 417.8 billion). Last year, the export value of domestically produced biopharmaceuticals to the U.S. grew to nearly match the import value. Last year, the U.S. imported 694.7 million dollars worth of biopharmaceuticals, a 7% increase from the previous year. The share of North America in the total exports of domestically produced biopharmaceuticals also expanded significantly over the past year. By continent, the North American region's share increased by 5 percentage points, from 14.7% in 2023 to 19.4% last year. The US ranked second in biopharmaceutical exports, following Hungary. Hungary's biopharmaceutical exports totaled USD 1.23346 billion, followed by Turkey (USD 432.06 million) and Brazil (USD 144.04 million). The analysis is that exports of biopharmaceuticals to the United States have been increasing in line with the growth of the domestic biosimilar industry. Biosimilars developed by domestic companies such as Celltrion and Samsung Bioepis are rapidly expanding their market share in the United States. Celltrion received four biosimilar approvals in the US this year alone, while Samsung Bioepis launched “Pyzchiva,” a biosimilar version of the autoimmune disease treatment “Stelara,” and ‘Epysqli,” a biosimilar version of the rare disease treatment “Soliris” in the US market this year. (Source:KoBIA) Top 10 biopharmaceutical export/import nations With exports of biopharmaceuticals to the U.S. on the rise, domestic companies are closely monitoring the Trump administration's tariff imposition. As the U.S. is a major export destination for Korean biopharmaceuticals, there are concerns that the imposition of tariffs will inevitably deal a blow to the domestic biopharmaceutical export industry. U.S. President Donald Trump recently announced plans to raise tariffs on certain pharmaceutical products by up to 250%. President Trump stated, “We will impose tariffs on certain pharmaceutical products soon, raise the tariff rate to 150% after one year, and then to 250% thereafter.” As the U.S. is set to announce the results of its Section 232 investigation under the Trade Expansion Act of 1962, the industry believes that the imposition of tariffs on pharmaceutical products is highly likely. Section 232 of the Trade Expansion Act allows the Department of Commerce to investigate the impact of imported goods on national security and enables the president to take appropriate measures. The US government launched an investigation into this matter in April. In line with this policy direction, domestic and foreign pharmaceutical and biotechnology companies have been accelerating their production investments in the US. AstraZeneca and Roche have each announced investments of USD 50 billion, Johnson & Johnson USD 55 billion, Eli Lilly USD 27 billion, Novartis USD 23 billion, and Sanofi at least USD 20 billion to expand their manufacturing capabilities in the US. Domestic companies such as Celltrion are also rushing to enter the U.S. market and secure production bases. Celltrion is reportedly in talks to acquire Eli Lilly's monoclonal antibody production plant in Branchburg, New Jersey. Earlier, Jung Jin Seo, chairman of Celltrion Group, stated that the company is pursuing the acquisition of a U.S. active pharmaceutical ingredient (API) plant to reduce uncertainties related to U.S. tariff risks.
- Company
- 'Vanrafia' for rare kidney disease receives ODD in Korea
- by Eo, Yun-Ho Aug 12, 2025 06:16am
- A new drug for rare kidney disease, 'Vanrafia,' has been granted orphan drug designation in Korea. The Ministry of Food and Drug Safety (MFDS) recently announced this through notificiation board. The indication for the designation is to 'treat adult patients with primary immunoglobulin A nephropathy (IgAN) with a urine protein-to-creatinine ratio (UPCR) over 1.5g/g.' Vanrafia (atrasentan) received accelerated approval from the FDA in April. This drug is a once-daily oral non-steroidal therapeutic agent that can be used in combination with supportive therapy, including a renin-angiotensin system inhibitor. It can also be used in combination with an SGLT-2 inhibitor. The efficacy of Vanrafia was proven through an interim analysis of the Phase 3 ALIGN study. The study has not yet proven whether Vanrafia can slow the decline of kidney function in patients with IgAN. However, in the ALIGN study, patients who received Vanrafia in combination with RAS inhibitor had a clinically and statistically significant 36.1% reduction in proteinuria compared to the placebo group. These results were observed as early as week 6 and were sustained for 36 weeks. The effect of Vanrafia on UPCR was consistent across all subgroups in the primary study cohort, including those with different baseline disease characteristics such as age, gender, race, eGFR, and proteinuria. Meanwhile, IgAN is a progressive, rare autoimmune kidney disease where the immune system attacks the kidneys, often causing glomerular inflammation and proteinuria. Up to 50% of IgAN patients with persistent proteinuria progress to kidney failure within 10-20 years of diagnosis, requiring maintenance dialysis and kidney transplantation. The response to treatment can be varied.
- Company
- US Biosecurity Act passing is ramping up
- by Kim, Jin-Gu Aug 12, 2025 06:14am
- The U.S. Congress is re-pursuing the Biosecurity Act, which failed to pass last year. The bill explains a comprehensive set of sanctions against companies designated as 'biotechnology companies of concern.' Analysis suggests that the bill's chances of passing have increased, as it addresses the procedural issues that were a stumbling block last year. The pharmaceutical industry anticipates that if this bill, which targets Chinese biotech companies, passes, Korean biotech companies could benefit from it. According to the Korea Biotechnology Industry Organization on August 11, Senators Bill Hagerty (Republican) and Gary Peters (Democrat) recently submitted an amendment to the National Defense Authorization Act to the Senate. This amendment incorporates the contents of the Biosecurity Act that were not passed last year. The core of the bill is similar to last year's version. The U.S. administration would be able to designate 'biotechnology companies of concern.' These designated companies would be restricted from federal procurement, contracts, loans, and grants within the U.S. Specifically, U.S. government agencies would be prohibited from procuring or acquiring biotech equipment and services from designated companies. They could not enter into, extend, or renew contracts for equipment and services produced or provided by these companies. Loans and grants could not be used to procure, acquire, or use equipment and services from these companies. However, the restrictions on equipment and services produced or provided under existing contracts would be deferred for five years. The Biosecurity Act was previously pursued by the U.S. Congress last year but failed. This was due to issues raised during the legislative process concerning the lack of transparency in the designation procedure for 'biotechnology companies of concern.' At the time, five Chinese biotech companies, including WuXi Biologics, were identified as targets for regulation, leading to criticism that the process of how they were specifically designated was unclear. The absence of a procedure for removing a company from the list of concern was also a point of criticism. The newly proposed bill aims to address these concerns. The bill explains that if a company is designated as a 'biotechnology company of concern,' the U.S. government must: ▲Notify the company of its designation ▲Provide the reasons for the designation to the extent consistent with national security and law enforcement interests ▲Allow the company to submit arguments opposing the designation within 90 days of receiving the notice ▲Explain the relevant rules and procedures ▲Inform the company about the process for rescinding the designation. These measures are being pursued not as a standalone bill but as an amendment to the NDAA. It would create a new 'SEC. 881' at the end of Title VIII, Subtitle E of the NDAA, specifically 'prohibiting contracts with certain biotech providers.' If the bill passes, the U.S. Office of Management and Budget (OMB) must publish a list of 'biotechnology companies of concern' within one year of the NDAA's enactment. The designated companies would be Chinese military companies operating in the U.S. that the Department of Defense publishes annually in the Federal Register. Also, the targets include: ▲Institutes that are subject to the administrative governance structure, direction, or control of, or are operated on behalf of, a government of a foreign adversary ▲Institutes involved in some capacity in the manufacturing, distribution, provision, or procurement of biotech equipment and services; and ▲Institutes that pose a risk to national security. The subsidiaries, parent companies, and affiliates of these entities would also be included. Industry analysis suggests that the bill's chances of passing are higher than last year's, as the procedure for designating and delisting 'biotechnology companies of concern' has been improved. It is also anticipated that the affected companies will mount a more intense backlash and lobbying effort. In the U.S., it is anticipated that the Senate could begin deliberating the bill as early as this September. If the bill passes, Korean biotech companies are expected to benefit. It is anticipated to be a significant opportunity for the overseas expansion of Korean Contract Development and Manufacturing Organization (CDMO) companies. However, concerns are also being raised that Korean companies could be negatively impacted, as many domestic firms currently collaborate with the Chinese companies in question. There is apprehension that companies working with firms like WuXi Biologics and WuXi AppTec could face disruptions to their business operations. Additionally, concerns have arisen that a strategy must be developed to differentiate Korea from other countries that also seek to fill the gap left by China. While China's absence presents a clear opportunity for Korea, the same holds for other countries, such as Japan and India. Consequently, they must consider ways to win the competition against these rivals.
- Company
- Gilead’s Livdelzi receives orphan drug designation in KOR
- by Eo, Yun-Ho Aug 11, 2025 06:05am
- The new primary biliary cholangitis drug ‘Livdelzi’ has received the orphan drug designation in Korea. The Ministry of Food and Drug Safety announced the news in a public notice on the 4th. Specifically, the designated indication is “the treatment of primary biliary cholangitis (PBC) in adults who have an inadequate response or intolerance to ursodeoxycholic acid.” Livdelzi (seladelpar) was also designated as a candidate for the Global Innovative Products on Fast Track (GIFT) program in June. PBC is a rare, intractable autoimmune disease characterized by chronic inflammation and destruction of the bile ducts in the liver, leading to bile stasis and liver damage, which can then potentially progress to cirrhosis and liver failure. While ursodeoxycholic acid (UDCA) is currently used as the first-line treatment, there has been an ongoing need for new treatment strategies for patients who have an inadequate response to UDCA or experience intolerance. Seladelpar is an oral selective peroxisome proliferator-activated receptor (PPAR) delta agonist. This drug demonstrated efficacy in a Phase III RESPONSE study. The RESPONSE study was a randomized, double-blind, placebo-controlled trial evaluating the efficacy and safety of seladelpar compared to placebo in adult PBC patients with inadequate response to UDCA or intolerance to UDCA. Study results showed that 62% of patients in the seladelpar treatment group achieved the primary endpoint (composite biochemical response, defined as improvement in alkaline phosphatase (ALP) levels and total bilirubin levels) at 12 months, demonstrating statistically significant superiority over the 20% in the placebo group. Notably, 25% of patients in the seladelpar treatment group achieved normalization of alkaline phosphatase (ALP) values, demonstrating significance. The efficacy of seladelpar was also confirmed in the improvement of pruritus, a key secondary endpoint. Compared to baseline, at 6 months, the seladelpar treatment group showed an average reduction of 3.2 points in pruritus scores among patients with moderate-to-severe pruritus, demonstrating a statistically significant improvement compared to the placebo group's reduction of 1.7 points. Meanwhile, Livdelzi was granted accelerated approval by the U.S. FDA in August last year and was approved in Europe in February this year.
- Company
- First TYK2i Sotyktu shows effect in Asian psoriasis patients
- by Son, Hyung Min Aug 11, 2025 06:04am
- BMS Korea The psoriatic arthritis treatment Sotyktu, which is jointly marketed in Korea by BMS Korea and Yuhan Corp, has established itself as a personalized oral treatment option for psoriatic arthritis patients in Korea, accumulating data from patients in East Asia. Sotyktu has been proven effective even in difficult-to-treat areas such as the scalp and nails, and is evaluated as having increased patients' access to early treatment through its inclusion in the national health insurance reimbursement list. According to industry sources on the 8th, BMS recently released clinical data of Sotyktu on East Asian patients, which included Koreans. Sotyktu is a first-in-class selective TYK2 inhibitor that, unlike other JAK inhibitors, has an allosteric mechanism that binds to the inactive regulatory domain. Psoriasis is a chronic disease that is difficult to cure and involves repeated improvements and worsening of symptoms, requiring long-term and continuous treatment. In particular, there remained a therapeutic need for new alternatives due to the diverse treatment options required based on individual disease characteristics and treatment preferences. Sotyktu is a once-daily oral treatment that can be taken regardless of meal intake and without dose adjustment, expanding the treatment options for psoriasis patients who previously had only biological agents as an alternative to conventional treatments. Sotyktu has demonstrated its efficacy and safety profile as a psoriasis treatment in numerous global and Asian clinical trials. In the POETYK PSO-3 study, which included Asian patients including Koreans, the proportion of patients who showed a 75% or greater reduction in the Psoriasis Area and Severity Index score (PASI 75) at Week 16 was 68.8%, and the rate of achieving a score of 0/1 on the static Physician’s Global Assessment (sPGA) was 55.6%, showing significant improvement compared to the placebo group. The results of three recent analyses conducted in Japan also supported the efficacy and safety of Sotyktu in East Asian patients. New data was released this year, including a post-hoc analysis of the POETYK PSO-4 Phase III clinical trial, a three-year efficacy and safety analysis of Japanese patients through the POETYK PSO-1, 4, and LTE studies, and an analysis of Japanese patient reports from POETYK PSO-4. In the post-analysis of POETYK PSO-4 in Japanese patients, the PASI 2 or less rate at week 16 of treatment in the Sotyktu group was 46.0%, and at week 52, the PASI 2 or less rate was 68.3% and the PASI 1 or less rate was 47.6%. In the patient-reported outcome (PRO) analysis of the same study, the rate of patients achieving a DLQI (Dermatology Life Quality Index) of 0/1 at week 52 reached 66.1%. Major symptoms such as erythema, scaling thickness, and severity of scaling began to improve from week 1 of treatment, with over 80% improvement by week 16, and approximately 90% of the effect was maintained or improved by week 52. Kiheon Jeong, Professor of Dermatology at Kyung Hee University Hospital (Insurance Director of the Korean Society for Psoriasis (KSP)), stated, “Areas such as the scalp and palms/soles, which are visible and cause daily discomfort, often have low patient satisfaction even with mild symptoms. We anticipate that oral medications with proven efficacy in these special areas will expand treatment options.” Health insurance coverage greatly reduces treatment costs and improves access to the latest treatments Sotyktu showed consistent efficacy even in patients with a variety of previous treatment histories, demonstrating its potential as a long-term treatment option. In a three-year post-hoc analysis including POETYK PSO-1, PSO-4, and long-term extension (LTE) studies, 65.6% of 125 Japanese patients had prior treatment experience, and 20.8% of these had a history of biological agent use. In the analysis of Japanese patients with PSO-1, the PASI achievement rate was 88.9% in the first year and remained at 87.5% in the third year, and the sPGA 0/1 response rate also showed a long-term response, from 74.1% in the first year to 66.7% in the third year. Similar results were observed in patients who switched from placebo to Sotyktu. This suggests that consistent clinical responses are possible over a long period of time, regardless of previous treatment experience. Sotyktu was approved in Korea in 2023 and was reimbursed the following year, greatly improving patient access and reducing their burden of cost compared to existing treatments. According to the health insurance reimbursement criteria, it can be administered to adult patients aged 18 years or older with chronic severe plaque psoriasis that has persisted for at least six months. Sotyktu was approved by the Ministry of Food and Drug Safety in 2023 and became eligible for reimbursement by health insurance eight months later. Reimbursement is available for adult patients aged 18 years or older with chronic severe plaque psoriasis that has persisted for six months or longer and who have ▲symptoms on BSA (body surface area) of 10% or more ▲PASI score of 10 or higher, and who have not responded to or cannot continue treatment with methotrexate or cyclosporine for at least three months due to efficacy or adverse effects, or have not responded to or cannot continue photochemotherapy or narrowband ultraviolet B therapy for at least three months due to efficacy or adverse effects. Professor Jeong said, “The accumulation of evidence from East Asian patients is encouraging for the psoriasis treatment environment in Korea. With reimbursement, the burden of treatment costs has been reduced, enabling quick and economical treatment even for patients who do not qualify for the special calculation benefits. This will provide practical treatment opportunities to more patients.”
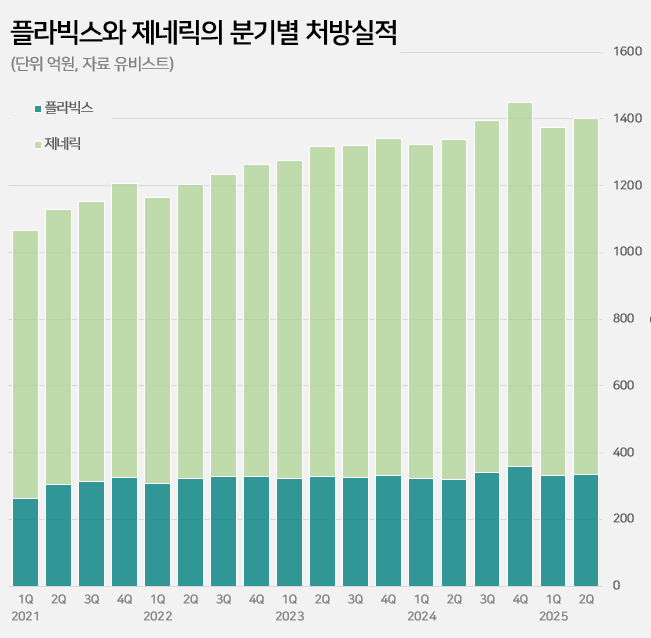
- Company
- Plavix’s sales continue to grow 27 years into its release
- by Kim, Jin-Gu Aug 11, 2025 06:04am
- Sales of the original drug ‘Plavix’ continue to rise in its 27th year of release in the clopidogrel-based antiplatelet agent market. It has maintained its lead with a 23% market share in the annual KRW 550 billion market. Meanwhile, major generic products are showing signs of slowing down. Samjin Pharmaceutical's ‘Platless’ saw a decrease in prescriptions compared to the same period last year, while sales of Dong-A ST's ‘Plavitor’ only increased by 2%. Sales of Chong Kun Dang’s ‘Pregrel' saw a 10% increase over the past year, but it still falls short of its performance before sales were suspended. Plavix’s sales in the first half of the year reach KRW 64.1 billion... Sales still on the rise even after 27 years on the market According to the pharmaceutical market research institute UBIST on the 9th, prescriptions for clopidogrel-containing antiplatelet drugs in the first half of this year reached KRW 264 billion, a 5% increase from the KRW 252.3 billion in the same period last year. The market leader is Handok’s Plavix. Sanofi launched Plavix in Korea in 1999. Since 2006, it has been manufactured by Handok. In 2016, a combination product called “Plavix A (Plavix+aspirin)” was added. Even after 27 years since its launch, sales of Plavix continue to grow in the outpatient prescription market. After the Plavix patent expired in 2007, 145 companies were approved to sell generic versions and entered the market. This is in contrast to the general trend of a decline observed in the prescription sales of original drugs after the launch of generic drugs. In fact, the combined prescription amount for Plavix and Plavix A exceeded KRW 100 billion for the first time in 2019 and continued to increase for 5 consecutive years thereafter. Based on the prescription amount rendered in the first half of the year, it increased by 29% in five years compared to KRW 49.8 billion in the first half of 2020. Quarterly prescriptions of Plavix and its generic versions The background behind Plavix's long-term growth lies in its market expansion strategy. Clopidogrel is mainly used to prevent the recurrence of ischemic cardiovascular diseases such as myocardial infarction and stroke, and has maintained its status as a standard therapy in domestic and international treatment guidelines along with an increase in the number of patients. The trust and prescription preference of medical professionals for the original drug have also contributed to its steady growth. Major generic products such as Samjin’s Platless and Dong-A ST's Plavitor show sluggish growth Meanwhile, sales of major generic products have recently shown signs of a slowdown. Samjin Pharmaceutical's Platless, the second-largest product in the market, recorded prescription sales of KRW 40.8 billion in the first half of this year. This represents a 4% decrease from the KRW 42.5 billion in the first half of last year. Dong-A ST's Plavitor, the third-largest product, saw a 2% increase in sales from KRW 15.3 billion to KRW 15.6 billion. Sales of Chong Kun Dang’s ‘Pregrel’ increased by 10% from KRW 11 billion to KRW 12.1 billion. However, it has yet to recover its pre-suspension sales performance. Pregrel’s sales were suspended from April to July 2021 after it was revealed that it had arbitrarily used additives. Before the suspension, Pregrel’s prescription sales exceeded KRW 7 billion every quarter, but since sales resumed, it has remained in the KRW 5 to 6 billion range. However, overall generic prescription sales increased by 4% from KRW 202.1 billion to KRW 210.7 billion in one year. This is believed to be the result of strong performance by products other than the top-ranked generics. Sales of Daewoong Pharmaceutical's 'Cloart' increased by 7% from KRW 10.4 billion to KRW 11.1 billion, and Hanmi Pharmaceutical's 'Pidogul' increased by 5% from KRW 9.7 billion to KRW 10.1 billion. Other products, such as JinYang Pharm’s Krivix (11%), Teragenix's Pravixen (14%), and Dasam Pharmaceutical's Clpigrel (54%), also recorded relatively high growth rates. Generic companies' clopidogrel-aspirin combination drugs are not faring so well in the market. HK Inno.N, Jeil Pharmaceutical, and Myungin Pharmaceutical have launched combination drugs containing the two ingredients since 2011. The combined prescription sales of these combination drugs decreased by 3% from KRW 13.8 billion in the first half of last year to KRW 13.4 billion in the first half of this year.

- Company
- The launch of Mounjaro is imminent
- by Moon, sung-ho Aug 11, 2025 06:03am
- The launch of Eli Lilly's 'Mounjaro,' which is called a 'miracle weight-loss drug' and garnered global attention alongside Wegovy, in Korea, is imminent. With the domestic launch of Mounjaro scheduled for mid-August, competition in the obesity treatment market, besides diabetes, is entering a new phase. There is significant interest in the sales and marketing strategies that Eli Lilly Korea will employ to challenge Novo Nordisk's Wegovy, which was launched in the domestic market approximately a year earlier. According to pharmaceutical industry sources on August 7, Eli Lilly Korea is set to launch the GIP/GLP-1 dual agonist Mounjaro Prefilled Pen Inj (tirzepatide), hereafter referred to as Mounjaro, in 2.5 mg and 5 mg dosages for patients with type 2 diabetes and obesity in mid-August. Mounjaro is the first and only GIP (Glucose-dependent Insulinotropic Polypeptide)/GLP-1 (Glucagon-like Peptide-1) receptor dual agonist. It is a single-molecule injectable designed to selectively bind to and activate GIP and GLP-1 receptors, allowing for once-weekly administration. It helps lower blood sugar by promoting insulin secretion, improving insulin sensitivity, and reducing glucagon concentration. Additionally, it aids in weight loss by delaying gastric emptying, which in turn reduces food intake. In Korea, Mounjaro is approved as an adjunct drug to diet and exercise for improving glycemic control in adult patients with Type 2 diabetes (as monotherapy or combination therapy). It is also approved as an adjunct to a low-calorie diet and increased physical activity for chronic weight management in obese adults (initial BMI≥30kg/m2) or overweight adults (initial BMI≥30kg/m2) with at least one weight-related comorbidity (hypertension, dyslipidemia, type 2 diabetes, obstructive sleep apnea, or cardiovascular disease). For both indications, the recommended starting dose is 2.5mg once a week (intended for treatment initiation and not for glycemic control or weight management). After four weeks, the dose is increased to 5mg once a week. If further dose adjustment is needed, the dose is increased by 2.5 mg after at least four weeks at the current dose, with a maximum dose of 15 mg once a week. Meanwhile, Eli Lilly Korea has decided to initially launch the 'pre-filled pen' formulation, which was first approved in 2023, amid delays in the approval of the vial and QuickPen formulations. This is interpreted as a strategy first to release the pre-filled pen formulation and subsequently launch the other formulations once their ongoing approval process is complete. Following the launch of Mounjaro, Eli Lilly Korea plans to implement a comprehensive sales and marketing strategy, which will include hosting symposium for medical professionals at major hospitals and clinics starting in late August. Consequently, the clinical community's attention is focused on Mounjaro's pricing. Since obesity treatment is not reimbursed, Mounjaro's supply price will be a critical factor. According to the pharmaceutical industry, Mounjaro is expected to be supplied at around KRW 278,000 for the 2.5mg dose (4PEN) and approximately KRW 369,000 for the 5mg dose (4PEN). This pricing is interpreted as a strategic move, considering that Novo Nordisk Pharma's Wegovy (semaglutide) was priced at KRW 372,025 per month for all dosages when it was launched in Korea last year. It's believed the 5mg dose was strategically priced to compete with Wegovy. Consequently, it's expected that doctors will also consider the drug's supply price when setting the average monthly price for non-reimbursed treatments. For reference, patients typically pay an average of KRW 500,000 to 600,000 per month for Wegovy as a non-reimbursed treatment at clinics. Given this, the non-reimbursed price for Mounjaro 2.5mg is likely to be lower than that of Wegovy, while the 5mg dose may be similar to Wegovy's monthly cost. Cheol Jin Lee, Chairman of the Korean Society for the Study of Obesity (Good Family Clinic), stated, "The vial and QuickPen formulations need approval, but it seems that the Ministry of Food and Drug Safety's (MFDS) approval is delayed." Lee added, "If the vial formulation is also launched in Korea, it would offer a significant pricing advantage. As far as we know, Mounjaro will initially be released in a pre-filled pen format, which is a disposable injectable form." Lee noted, "Since the efficacy of this drug was confirmed in clinical trials, if it is launched, it will likely be used primarily for new patients." He explained, "This is because it would be difficult for patients who are already on high-dose Wegovy to switch to Mounjaro, which starts at a 2.5mg dose. When considering efficacy and other factors, we would likely consider Mounjaro for new patients if it is launched." With Mounjaro's launch, the focus shifts to its competition with Wegovy in clinical practice. Novo Nordisk is also actively ramping up its sales and marketing efforts as the first anniversary of Wegovy's domestic launch approaches. In addition to its in-house sales team of around 80 people, the company is reportedly considering a joint sales and marketing strategy with a domestic pharmaceutical company. Chong Kun Dang is a strong candidate being discussed within the pharmaceutical industry as a domestic sales partner. However, Novo Nordisk's position is that while they are pursuing collaboration with a domestic pharmaceutical company, nothing has been finalized. At the same time, Novo Nordisk is also seeking to expand Wegovy's indication to adolescents aged 12 and older. If this becomes a reality, it would give Wegovy a differentiated competitive edge over Mounjaro in domestic clinical practice, primarily because adolescent obesity has emerged as a significant social issue. Eli Lilly Korea has also established its own sales force, and like Novo Nordisk, is open to collaboration with domestic pharmaceutical companies. Although several companies, including Hanmi Pharm, are being mentioned, Lilly is expected to focus on Mounjaro's launch for the time being. In this context, the pharmaceutical industry is also closely monitoring Eli Lilly Korea's efforts to secure reimbursement for Mounjaro's diabetes indication. There is great interest in whether it will be recognized as an innovative new drug, alongside its competitor, Ozempic. This contrasts with Novo Nordisk's strategy, which involves applying different indications for a single product as part of its reimbursement strategy. An official said, "We have applied to the Health Insurance Review & Assessment Service (HIRA) for national health insurance coverage and are currently awaiting consideration from the DREC." They added, "As the first GIP/GLP-1 receptor dual agonist and a new treatment for type 2 diabetes, we expect Mounjaro to provide differentiated clinical value. Therefore, we are doing our best for it to be the first chronic disease drug to receive flexible application of the ICER value for innovative new drugs." A pharmaceutical industry official, who requested anonymity, expressed doubt about whether Mounjaro could meet the government's definition of innovation. They said, "Of course, this doesn't mean Mounjaro has no innovative value. However, we need to verify whether it meets the criteria and definition of innovativeness that the government has established. Ultimately, the question of whether it can be applied will inevitably follow." An official also said, "We are curious about how the drug price will be set if one treatment is covered for diabetes but not for obesity," and added, "With Ozempic also pursuing reimbursement with a target of the first half of next year, this issue will become a new topic of discussion."

- Company
- New pulmonary hypertension drugs are competing for approval
- by Moon, sung-ho Aug 11, 2025 06:03am
- Global pharmaceutical companies are introducing new treatment options for pulmonary hypertension, utilizing novel mechanisms, which are being successively approved in the Korean market. These drugs are expected to change the clinical treatment paradigm. It's anticipated that they will establish a new market alongside the currently reimbursed treatments. According to the pharmaceutical industry source on July 28, the Ministry of Food and Drug Safety (MFDS) recently approved Winrevair (sotatercept) from MSD Korea and Opsynvi Tab (macitentan-tadalafil) from Janssen Korea as treatments for pulmonary hypertension. Pulmonary hypertension is a rare, severe, intractable disease where the walls of the pulmonary arterioles abnormally thicken and narrow, causing pressure to rise. As the disease progresses, symptoms like shortness of breath, chest pain, and fainting appear, severely limiting all aspects of daily life. Additionally, the increased pressure in the right ventricle due to the narrowed pulmonary arteries gradually weakens the function of the right side of the heart, leading to a high risk of sudden death. First, MSD Korea's Winrevair is an 'Activin Signaling Inhibitor (ASI), the first-in-class (as of July 2025)' to be approved in the field of pulmonary hypertension. It is a new treatment mechanism that has emerged after 20 years. Its mechanism directly addresses the fundamental cause of the disease by blocking the excessive proliferative signals of the protein complex 'activin,' which causes cell proliferation within pulmonary artery blood vessels, and by restoring the balance with anti-proliferative signals that inhibit cell growth, thereby inducing reverse remodeling to normalize the deformed vascular structure. MFDS approved Winrevair for use in combination with existing treatments to improve the exercise capacity of adult patients (18 years or older) with pulmonary hypertension (WHO Group I) who are in WHO Functional Class II-III. The three existing treatments are endothelin receptor antagonists (ERA), PDE-5 inhibitors (PDE-5i), and prostacyclin analogues (PCA). With the addition of this new mechanism, the Activin Signaling Inhibitor, which differs from existing treatments, has further broadened the options for pulmonary hypertension patients. The STELLAR clinical trial, which served as the basis for approval, evaluated the efficacy and safety of Winrevair in 323 adult patients with pulmonary hypertension in WHO-FC II or III. During the 24-week study period, patients received either Winrevair or a placebo in combination with their existing treatment once every three weeks. Winrevair increased the 6-minute walk distance by 40.8m compared to placebo at the 24-week mark (95% CI, 27.5-54.1; P
- Company
- Jaqbo’s substance patent extended to 2040
- by Kim, Jin-Gu Aug 08, 2025 06:03am
- Onconic Therapeutics announced on the 7th that the patent term for Jaqbo (zastaprazan), a new drug for gastroesophageal reflux disease in the P-CAB (potassium-competitive acid blocker class, has been extended by four years and two months. According to the company, the Korean Intellectual Property Office recently extended the patent term of Jaqbo’s substance patent, “Imidazo[1,2-a]pyridine derivatives, methods for preparing the same and use thereof” from July 5, 2036, to September 13, 2040, for approximately 4 years and 2 months. The KIPO recently made an official decision on the extension registration and published it in the official gazette. The patent term extension registration system is designed to compensate for the reduction in the actual patent term due to issues such as the time taken for marketing authorizations. If certain requirements are met, the patent term can be extended for up to five years, which is considered a key mechanism for protecting the intellectual property of new drug development companies and securing market competitiveness. Jaqbo Onconic Therapeutics’ new drug, which was approved in April last year as the 37th domestically developed new drug. It is a P-CAB gastric acid secretion inhibitor and is evaluated to have faster efficacy and superior nighttime gastric acid control compared to existing PPI (proton pump inhibitor) type treatments. Recently, it has been approved for use in treating not only gastroesophageal reflux disease but also gastric ulcers. Jaqbo’s sales surpassed KRW 10 billion in quarterly prescriptions less than a year after its launch in October last year. The company expects that the patent extension will accelerate sales growth in line with its strategy to expand the indications for Jaqbo and advance into global markets. An official from Onconic Therapeutics said, “With this patent extension, Jaqbo will be able to maintain its exclusive position in the domestic market until 2040,” adding, “As the rights to our independently developed new drug have been strengthened, we plan to focus our capabilities on the development of subsequent pipelines.”