- LOGIN
- MemberShip
- 2026-04-28 01:31:10

- Company
- "6-in-1 vaccine Hexaxim may enhance preventive effect of NIP
- by Whang, byung-woo Jun 23, 2025 06:00am
- "Hexaxim is Korea's first hexavalent vaccine. It is expected to play a crucial role in helping protect children's health." Following the inclusion of Hexaxim, a hexavalent (6-in-1) DTaP vaccine, in the National Immunization Program (NIP) this year, the medical field is holding high hopes for increased convenience of immunization and improvement in vaccination rates. The analysis is that since immunization through NIP has already been conducted, the field is already experiencing practical benefits, such as a reduced number of immunizations. It is believed that, in the long term, Hexaxim will lessen the burden of hospital visits for caretakers, alleviate the stress of infant injections, and prevent the skipping of immunizations. Dr. Byung Wook Eun, Professor of Department of Pediatrics at Nowon Eulji Medical CenterDaily Pharm met with Dr. Byung Wook Eun, a professor at Nowon Eulji Medical Center, and listened to the background of the introduction of Hexaxim and its potential effects. Hexaxim is Korea's first and only fully liquid hexavalent fully liquid formulation vaccine, preventing six diseases, including diphtheria, tetanus, pertussis (DTaP), inactivated polio vaccine (IPV), Haemophilus influenzae type b (Hib), and hepatitis B (HepB), with a single injection. As of January 2, it has been officially introduced into the NIP, allowing the previous separate administration of the pentavalent (5-in-1) vaccine (DTaP-IPV-Hib) and the monovalent Hepatitis B vaccine to now be given in a single shot. According to Dr. Eun, since the introduction of Hexaxim into the NIP, there has been an increase in inquiries regarding new vaccination schedules and methods in actual clinical settings. Dr. Eun said, "With the introduction of the hexavalent vaccine, the vaccination schedule differs from the existing pentavalent vaccine, leading to many questions from both guardians and medical staff regarding vaccination criteria, interchangeability, and delayed vaccinations." Hexaxim is already a proven vaccine overseas, having received its first approval in Europe in 2013. It is now used in over 120 countries worldwide and is recommended as a mandatory vaccination in more than 40 countries, including Europe, Canada, and Australia. Dr. Eun explained, "The immunogenicity and safety of Hexaxim against all six infectious diseases through global clinical studies, including those conducted in Korea," and added, "In Korea, non-inferiority related to immunogenicity was confirmed through a clinical trial comparing the Hexaxim vaccination group with the group receiving the existing pentavalent DTaP combination vaccine concurrently with the monovalent Hepatitis B vaccine." Fewer injections due to schedule changes help improve convenience The introduction of Hexaxim has brought significant changes to the infant vaccination schedule. Previously, infants needed to receive the Hepatitis B vaccine at 0, 1, and 6 months of age and the pentavalent DTaP vaccine at 2, 4, and 6 months. However, starting this year, the schedule has changed. After the first Hepatitis B vaccination immediately after birth, infants will now receive Hexaxim at 2, 4, and 6 months, with no additional vaccination at 1 month. Dr. Eun stressed that "The introduction of Hexaxim reduces the total number of vaccinations by two, bringing the total to four injections." He added, "The hexavalent DTaP vaccine can administer six antigens at once, which can help improve vaccination rates compared to administering individual vaccines separately." However, as with any new vaccine introduction, there's some initial confusion in the field. Dr. Eun explained that there have been inquiries regarding the switching to the hexavalent vaccine at 6 months if the infant has already received the pentavalent combination vaccine at 2 or 4 months. The Korea Disease Control and Prevention Agency (KDCA) is addressing this confusion with clear guidelines. Dr. Eun stressed, "According to KDCA's official guidelines, DTaP-containing vaccines vary by manufacturer. Therefore, for the primary 3-dose series, it is recommended to administer vaccines from the same manufacturer consistently." Dr. Eun also said, "However, since most newborns are currently starting their vaccinations with Hexaxim, inquiries about interchangeability will gradually decrease in the future," and added, "There are also questions about vaccination age and intervals. Hexaxim's recommended vaccination schedule is at 2, 4, and 6 months of age (8-week intervals), with the recommended age for the first dose being 8 weeks (2 months of age)." However, in unavoidable circumstances, the first dose can be administered as early as 6 weeks of age. While a 2-month (8-week) interval is generally recommended, the vaccination schedule can be adjusted to a minimum of 4-week intervals. For example, if the first dose was given at 3 months of age, the second dose can be given 4 weeks later. Dr. Eun stated, "If there are any changes to the vaccination schedule, it is crucial to consult with a healthcare professional to get guidance on the timing and vaccine selection." There are, however, exceptional cases where Hexaxim cannot be administered. For instance, newborns born to mothers who are positive for the Hepatitis B virus must receive Hepatitis B immunoglobulin and the Hepatitis B vaccine within 12 hours of birth. These high-risk infants also require additional Hepatitis B vaccine doses at 1, 2, and 6 months of age, thus excluding them from the Hexaxim program. Furthermore, infants who have already completed their second Hepatitis B vaccination by 1 month of age cannot switch to Hexaxim and must continue their remaining vaccinations with the existing combination of the pentavalent vaccine and the Hepatitis B vaccine. "Hexaxim with increased preventive effects... high hopes for increasing vaccination rates" Dr. Eun stated that concerns about Hepatitis B antibody formation, which were previously raised by some, have been addressed through research. In the previous schedule, Hepatitis B vaccine was administered at 0 and 1 months of age. However, with the Hexaxim schedule, the 1-month vaccination is omitted, changing the schedule to 0-2-4~6 months. Regarding this, Dr. Eun responded, "The 0-2-4~6 month Hepatitis B vaccination schedule using Hexaxim showed immunogenicity similar to the existing 'pentavalent vaccine+Hepatitis B vaccine' schedule," and added, "Although the hexavalent vaccine includes a Hepatitis B component in addition to the existing pentavalent combination vaccine, its safety profile was confirmed to be similar to separate administrations." One of the anticipated effects of including Hexaxim in the NIP is an improvement in vaccination rates. Dr. Eun said, "Because a hexavalent vaccine can introduce six antigens in one shot, it can help improve vaccination rates compared to administering individual vaccines separately," and added, "Currently, infant primary vaccination rates exceed 90%, but we expect to push this figure higher with Hexaxim's introduction." As mentioned earlier, the reduced number of vaccinations helps prevent delayed vaccinations, which in turn can lead to a higher 'right-time vaccination rate,' thereby completing vaccinations at the recommended period, thus improving overall vaccination rates. Indeed, improvements in vaccination rates have been observed in other countries after the introduction of hexavalent combination vaccines. In Australia, after the hexavalent vaccine was included in their NIP, the vaccination rate for infants aged 12 months increased from 84.9% in 2009 to 92.6% in 2018. Lastly, Dr. Eun added, "Misinformation about vaccines can lead to a drop in vaccination rates, and previously well-managed infectious diseases could potentially re-emerge," and urged, "We hope the public would utilize the NIP to help protect children's health."
- Company
- Cream JAKi Delgocitinib to be commercialized in Korea
- by Eo, Yun-Ho Jun 20, 2025 06:06am
- JAK inhibitors will soon be available for hand eczema in Korea as well. The Ministry of Food and Drug Safety is currently conducting a final review of LEO Pharma Korea's treatment for moderate-to-severe chronic hand eczema (CHE), Anzupgo (delgocitinib). When approved, the drug is expected to be officially commercialized in the second half of the year in Korea. Anzupgo was officially approved in Europe in November last year and is currently awaiting approval from the US FDA. This cream formulation is known as a pan-JAK inhibitor, as it targets JAK1, 2, and 3, as well as TKY2. The efficacy of Anzupgo has been demonstrated through the DELTA FORCE and DELTA 2 clinical trials, which directly compared Anzupgo with GSK's alitretinoin (Toctino). In the DELTA FORCE study, the Hand Eczema Severity Index (HECSI) score from baseline to week 12 of delgocitinib demonstrated superiority over alitretinoin capsule, meeting the primary endpoint. The DELTA 2 study included 473 patients with moderate-to-severe chronic hand eczema (CHE). Study participants were assigned to either the delgocitinib cream group or the placebo cream group and received treatment twice daily for 16 weeks. The primary endpoint was set as an IGA-CHE score of 0/1 at Week 16 of treatment. The primary secondary endpoints included IGA-CHE and the Hand Eczema Symptom Diary (HESD) assessed at Weeks 4 and 8 of treatment. The results showed that the delgocitinib group achieved significant improvement in chronic hand eczema at Week 16 compared to the placebo group, meeting the primary and primary secondary endpoints. Chronic hand eczema can be triggered by various factors such as irritants, allergies, genetic predisposition, and occupational exposure. It often leads to recurring inflammation, peeling, cracking, and pain, significantly impacting daily life. However, to date, effective topical treatments specifically targeting hand eczema remain limited. In many cases, patients show insufficient response even to corticosteroids or immunosuppressants, underscoring the ongoing need for new therapeutic alternatives. Meanwhile, LEO Pharma obtained exclusive rights in 2014 from the Japanese company Japan Tobacco to develop and commercialize a topical delgocitinib cream for dermatological indications worldwide, excluding Japan.
- Company
- Interest in Lotte Biologcs’ partner Ottimo Pharma rises
- by Kim, Jin-Gu Jun 20, 2025 06:05am
- Interest is growing in Ottimo Pharma, a biotech company that has signed a contract manufacturing agreement for its antibody-drug with Lotte Biologics. Founded in 2017, this UK-based biotech venture owns a new drug candidate called ‘Jankistomig’. The drug candidate bifunctional antibody targeting solid tumors, and the company plans to submit an Investigational New Drug (IND) application to the US Food and Drug Administration (FDA) within this year. Lotte Biologics announced on the 19th that it has signed an antibody-drug contract manufacturing agreement with Ottimo Pharma at the BIO INTERNATIONAL 2025 (Bio USA) event. The contract covers the production of drug substance (DS) for Ottimo Pharma's antibody drug Jankistomig at the Syracuse Bio Campus in New York. Ottimo Pharma was founded in 2017 in Kent, England, under the name Ultrahuman Eight Limited. In October last year, the company changed its name to Ottimo Pharma. At that time, Medicxi Ventures UK, a UK-based life science venture capital, participated as an initial investor. Medicxi led Ottimo Pharma's Series A investment round. Ottimo Pharma successfully secured USD 140 million (approximately KRW 190 billion) in Series A investment in December last year. To date, Ottimo Pharma’s only pipeline is Jankistomig. This candidate drug works by simultaneously inhibiting PD-1 and VEGFR2. The drug candidate was known to be designed based on camrelizumab, developed by China's Jiangsu Hengrui Pharmaceuticals. It is designed to reduce VEGF-related side effects while providing immune checkpoint inhibition effects. Jankistomig is in the preclinical stage and is being tested in the UK for solid tumors. There is no clinical trial number registered on ClinicalTrials.gov, a clinical trial registration site. This suggests that no official clinical trials have been initiated in any country, including the US and the UK. The company announced last year that it had secured Series A investment and would submit an Investigational New Drug (IND) application to the US FDA by the end of this year. At the time, Ottimo mentioned its development of other pipelines in addition to Jankistomig, but did not disclose specific substance names or stages.

- Company
- New pneumococcal vaccine expected to be launched
- by Whang, byung-woo Jun 19, 2025 06:04am
- As 'Capvaxive,' a 21-valent pneumococcal conjugate vaccine (PCV21) developed by MSD, is anticipated to receive marketing authorization in South Korea, competition in the market is likely to heat up. Product photo of CapvaxiveAccording to pharmaceutical industry sources, MSD has filed with the Ministry of Food and Drug Safety (MFDS) for marketing authorization of Capvaxive. It is expected to be approved by the second half of 2025. Capvaxive is a vaccine designed to prevent adults from serotype that causes most of the invasive pneumococcal disease (IPD). The safety and immunogenicity of Capvaxive were cofirnmed based on the Phase 3 STRIDE clinical trial, comparing Capvaxive to PCV20 in adults aged 18 years and above who have no prior history of pneumococcal vaccination. Capvaxive was found to be nonequivalent to PCV20 regarding 10 serotypes (3, 6A, 7F, 8, 10A, 11A, 12F, 19A, 22F, 33F) that are commonly included in PCV20. 10 out of 11 serotypes (9N, 15A, 16F, 17F, 20A, 23A, 23B, 24F, 31, 35B) that are included in Capvaxive but not in PCV20 were demonstrated to be superior to PCV20. Capvaxive was approved in the United States in June 2024 based on these study results, and it also obtained European approval in March. There is growing attention on whether Capvaxive will obtain Korean approval during the second half of this year, as it will be the third consecutive year a new pneumococcal vaccine is approved in South Korea. In late 2023, MSD's 15-valent vaccine, 'Vaxneuvance,' was expedited for inclusion in the pediatric National Immunization Program (NIP). Then, a year later, in October 2024, Pfizer's 20-valent vaccine, Prevenar 20, won MFDS approval. As the 21-valent vaccine, which is the higher serotype vaccine, is expected to be introduced in less than a year, competition is likely to get intense. If there are no setbacks to the approval process for Capvaxive, it is expected to be launched at the very end of the first half of next year. For instance, Vaxneuvance was launched in late April, and Prevenar 20 was launched in June exclusively for adults aged 18 years and above. The market is also highly likely to be competitive, with Vaxneuvance and Prevenar 20 competing for the pediatric NIP and Prevenar 20 and Capvaxive competing for the adult NIP. Regarding this, MSD Korea is expected to employ a marketing strategy that differentiates its vaccine portfolio for pediatric (15-valent) and adult (21-valent) populations. Indeed, MSD previously announced the use of tailored strategies for pediatric and adult populations during its Vaxneuvance launch 1st-anniversary media seminar. In the long term, Pfizer's Prevenar 20, with its first-mover advantage, will be competing directly with MSD's 21-valent Capvaxive, which includes a higher number of serotypes. Capvaxive will reportedly be preventing approximately 84-85% of adult IPD. This estimate is higher than the coverage for Prevenar 20. In this case, Pfizer is expected to defend its position by highlighting the performance and extensive clinical experience of Prevenar 20. Currently, Pfizer emphasizes that Prevenar 20 offers safety and convenience based on the well-established technology of Prevenar 13, validated through long-term pediatric and adult vaccinations, and with its 20-serotype coverage. Additionally, potential competition against Sanofi-SK bioscience is another variable. Although their commercialization timeline is the latest, if they succeed in developing a 21-valent vaccine, another equally strong competitor will emerge. Notably, the Sanofi vaccine is being developed for pediatric use, suggesting that the company will employ a future strategy to cover all age groups, from infants and young children to adults.

- Company
- K-Bios face string of clinical failures in H1
- by Son, Hyung Min Jun 19, 2025 06:04am
- A series of clinical trial failures for new drug candidates under development by Korean biotech companies in the first half of the year have raised concerns about the feasibility of future technology exports. Orum Therapeutics halted a clinical trial due to safety concerns, while Genexine and Bridge Biotherapeutics both failed to demonstrate statistical significance in their respective Phase II trials for glioblastoma and idiopathic pulmonary fibrosis. Stem cell therapy developers such as Anterogen and SCM LifeScience are also struggling to prove efficacy in clinical settings. According to industry sources on the 19th, Orum Therapeutics recently suspended its Phase 1 trial of ORM-5029. ORM-5029 was the company’s only pipeline in clinical trials targeting human epidermal growth factor receptor 2 (HER2), a major biomarker for solid tumors. The company received IND approval for ORM-5029 from the U.S. FDA in 2022. However, a severe adverse event (sAE) occurred during the trial, upon which the company reported it to the FDA. Due to toxicity issues, administration had to be halted even at low doses. ORM-5029 is a Degrader Antibody Conjugate (DAC) candidate. DACs combine Targeted Protein Degradation (TPD) mechanisms with Antibody Drug Conjugates (ADCs) and are expected to offer higher safety due to the use of TPD, which are small-molecule degraders. Orum emphasized that the sAE was limited to only the ORM-5029 substance and that there were no issues with the company's technology or platform itself. Orum plans to focus its resources on its blood cancer candidate ORM-1153, which also utilizes the company’s DAC platform. The company explained that it has shown strong GSPT1 degradation and robust anti-proliferative effects in blood cancer cell lines. Genexine and Bridge Biotherapeutics fail Phase II trials Genexine and Bridge Biotherapeutics both faced setbacks in Phase II trials. In March, Genexine's GX-I7 (Interleukin-7), an immune-oncology drug candidate, failed to demonstrate efficacy in glioblastoma mulifrome (GBM) patients. GX-I7 is a new drug candidate that maximizes immune anticancer effects by inducing T-cell amplification in the body. GBM is a type of glioma, a malignant tumor that originates in the brain. Despite surgery and chemotherapy, the five-year survival rate for GBM is only 5%, with an average survival time of about a year. The Phase II trial for GX-I7 enrolled 20 patients with recurrent or progressive glioblastoma, and evaluated a combination of the GX-I7 and bevacizumab (Avastin), a VEGF inhibitor used as a targeted therapy. Bevacizumab inhibits angiogenesis to prevent tumor growth, and its combination with existing anticancer drugs was expected to enhance therapeutic efficacy. However, no significant improvement was observed in the primary endpoints of progression-free survival (PFS) and overall survival (OS). Meanwhile, Bridge Biotherapeutics announced in April that its top-line data analysis results showed that its idiopathic pulmonary fibrosis (IPF) candidate BBT-877 failed to demonstrate a statistically significant improvement in the primary endpoint of forced vital capacity (FVC) change at 24 weeks. BBT-877 is an innovative novel drug candidate that selectively inhibits the novel target protein autotaxin. Autotaxin is a protein known to bind to intracellular receptors and be involved in pathological mechanisms such as fibrosis and tumorigenesis. BridgeBio previously secured global exclusive rights to BBT-877 from LegoChem Bio (now LigaChem Bio) in 2017. In May, BridgeBio received a recommendation from the IDMC to continue the clinical trial. The Phase 2 clinical trial of BBT-877 was conducted in 5 countries - South Korea, the United States, Australia, Poland, and Israel - to evaluate the efficacy, safety, and tolerability of the drug in patients with idiopathic pulmonary fibrosis (IPF). A total of 129 patients participated, and the study results showed that changes in FVC were observed in both the drug group and the placebo group; however, there was no statistically significant difference between the two groups. Bridge Biotherapeutics licensed out BBT-877 to Boehringer Ingelheim in 2019 in a deal worth up to KRW 1.5 trillion. Upon transferring BBT-877, which was in Phase 1 clinical trials, the company received approximately KRW 600 billion in upfront and milestone payments (short-term milestones). In late 2019, following the completion of Phase I clinical trials for BBT-877, BridgeBio paid approximately KRW 50 billion to LigaChem Bio as milestone revenue sharing. However, in 2020, Boehringer Ingelheim returned the rights to BBT-877 due to potential toxicity issues. BridgeBio determined that the toxicity issues were caused by high-dose drug administration in additional experiments and decided to develop the candidate on its own, but failed to demonstrate its efficacy in trials. stem cell therapy developers also struggling with commercialization Stem cell therapy developers are also facing commercialization hurdles. SCM LifeScience failed to achieve statistical significance in Phase 2 clinical trials of its stem cell therapy candidate SCM-CGH. This is the company's second failed attempt at commercialization following the failure of its acute pancreatitis clinical trial in 2022. The trial, which targeted patients with steroid-resistant or steroid-dependent chronic graft-versus-host disease, was conducted from 2017 to 2024 at 11 hospitals in South Korea, including Seoul St. Mary's Hospital. The results of the Phase II clinical trial of SCM-CGH showed no statistically significant difference in the primary efficacy endpoint, the overall response rate (ORR) at 12 weeks. Upon closer examination, the ORR at 12 weeks was higher in the placebo group than in the SCM-CGH group, and the results were not statistically significant. Anterogen failed to demonstrate the efficacy of its stem cell therapy ALLO-ASC-DFU in the U.S. Phase III clinical trial. In the trial, ALLO-ASC-DFU recorded a complete wound closure rate of 46%, which was lower than the 60% in the control group that was treated with hydrogel sheets. The therapy had garnered attention as a treatment for diabetic foot ulcers (DFU), but its failure to meet the key primary endpoint has significantly reduced the likelihood of its FDA approval. Anterogen is conducting further analyses to revise its development strategy.
- Company
- KOR-JPN jointly launches Healthcare Distribution Alliance
- by Son, Hyung Min Jun 19, 2025 06:03am
- (From the left) Jun-Jae Hyeon (CEO, Dongwon Healthcare), Jun-ho Hyun (CEO, Dongwon Pharmaceutical Wholesale), Seung-Uk Eom (CEO, Boksan Nice), and Seongwook Cho (Country Manager, Suzuken Korea) Three pharmaceutical distribution companies in Korea and Japan have joined forces to launch the Healthcare Distribution Alliance to lead the domestic market by introducing advanced overseas models. The alliance aims to go beyond simple logistics agreements – it seeks to build an innovative cooperation structure where companies can share capital and operational know-how, and combine each company's strengths to transform the pharmaceutical distribution market. Jun-Jae Hyeon (CEO, Dongwon Healthcare), Jun-ho Hyun (CEO, Dongwon Pharmaceutical Wholesale), Seung-Uk Eom (CEO, Boksan Nice), and Seongwook Cho (Country Manager, Suzuken Korea) recently met with reporters to explain the alliance's goals. Eight affiliates of Dongwon Pharmaceutical Group, Boksan Nice, and Suzuken have signed a business partnership agreement and established an organizational framework for cooperative operations at the alliance level. As part of the collaboration, the companies also entered into a capital partnership, with Suzuken acquiring a 33.6% stake in Gyeongnam Dongwon Pharmaceutical, and Boksan Nice acquiring a 3.4% stake in Gyeongnam Dongwon Pharmaceutical. This alliance goes beyond simple logistics cooperation by sharing capital and strategy direction of the companies. With the direct participation and investment of Suzuken, a major Japanese pharmaceutical distribution company, the alliance aims to pursue a long-term model that pursues the maximization of distribution productivity, supply chain stability, and function as part of a social infrastructure. CEO Seung-Uk Eom said, “We decided to pursue this alliance to survive in the rapidly changing pharmaceutical distribution industry and create growth opportunities through innovation. We aim to realize economies of scale through the alliance between Dongwon Pharmaceutical, Boksan Nice, and Suzuken and maximize productivity in the pharmaceutical distribution market while driving innovation for mutual growth.” Industry observers expect synergy from the partnership. Dongwon Pharmaceutical Group and Boksan Nice each reported over KRW 1 trillion in annual sales last year. Suzuken, one of Japan's top three pharmaceutical distributors, posted annual revenue exceeding JPY 2 trillion (approx. KRW 19 trillion) in 2023. CEO Jun-ho Hyun emphasized, “As the pharmaceutical distribution environment evolves and capital requirements grow, scaling up is no longer an option – it’s a necessity. We aim to establish a Korean-style large-scale distribution model and guide the future direction of the market.” The alliance anticipates increased distribution-related costs and volatility in the coming years. To address this, it aims to build a robust infrastructure and reduce labor dependency. Plans include exploring hospital market strategies, logistics outsourcing services, private-label (PB) healthcare products, and the potential introduction of Suzuken’s current Japanese business operations into the Korean market. CEO Seung-Uk Eom noted, “In the short term, we’ll focus on collaboration between logistics centers within the alliance, which is expected to reduce stockouts and delivery lead times through optimal inventory and shipping operations.” He added, “In the long term, we plan to build systems such as enterprise resource planning (ERP), web order systems (WOS), and customer relationship management (CRM). Given the significant time and cost required for IT system development, combining the long-standing expertise and ideas of Suzuken, Boksan Nice, and Dongwon Pharmaceutical will not only facilitate joint development but also greatly contribute to future logistics innovations such as the modernization of logistics and improvements in operational efficiency involving robots and AI. CEO Seung-Uk Eom added, “Beyond transportation management systems (TMS) and quality control standards, we will also build a foundation system for environmental, social, and governance (ESG) and seek ways to advance them.” CEO Jun-ho Hyun said, “Profit margins for pharmaceutical distributors have been steadily shrinking. Survival through sales promotion activities alone is becoming difficult. We must scale up and differentiate through cost reduction and pharmaceutical partnerships.” ”Will seek to implement Japanese-style wholesale structure in Korea" The alliance is eyeing the Japanese model, where pharmaceutical distribution is treated as a core part of national infrastructure, with government, pharma companies, wholesalers, and hospitals working in unison. Even logistics center placements are coordinated with government authorities, and disaster response systems are embedded into the distribution network. Japan’s market is dominated by major distributors like Medipal, Alfresa, and Suzuken, which fulfill roles as social infrastructure through close cooperation across the pharmaceutical supply chain. CEO Jun-jae Hyeon noted, “In Japan, systems are in place to ensure medicine continues to flow even during national disasters like earthquakes and tsunamis. We aspire to build such a socially integrated distribution system here in Korea.” Country Manager Seongwook Cho said, “, “Japan has established a virtuous cycle model that contributes to the national health insurance budget by minimizing the deterioration of medicine quality and the amount of expired medicines through infrastructure development. Although this may not be immediately achievable in South Korea, we will do our best to prepare for it." In addition, as more and more pharmaceutical companies are expected to develop new drugs such as biological agents, anticancer drugs, and orphan drugs, it is necessary to establish a system to manage and deliver these drugs. The association aims to provide one-stop services tailored to their needs. Country Manager Seongwook Cho said, “Suzuken already communicates and conducts business with many multinational pharmaceutical companies in Japan. We are aware that the companies have high standards for quality control and other global requirements. Our association’s goal is to meet the standards set by such global companies in various areas, including logistics and cold chain.” CEO Jun-jae Hyeon said, “In Japan, there are various pharmaceutical platforms, with distribution companies at the center of each. All transactions between healthcare institutions and related organizations are conducted through distribution companies. We will strive to establish a similar structure in Korea, where distribution companies play a central role in facilitating various activities.”
- Company
- Takeda launches new drug 'Fruzaqla' in Korea
- by Whang, byung-woo Jun 18, 2025 10:28am
- Product photo of Takeda Pharmaceutical's Fruzaqla Takeda Pharmaceutical Korea (CEO Kwang-kyu Park) announced on June 16 that the company has officially launched its 'Fruzaqla (fruquintinib)' in South Korea. Fruzaqla is the first new treatment for metastatic colorectal cancer. This drug selectively inhibits Vascular Endothelial Growth Factor Receptor (VEGFR)-1,2, and 3. It is expected to provide a new treatment option for patients subjecting to fourth-line or later treatment who had limited treatment options previously. According to the 2024 statistics, colorectal cancer is one of the cancer types with prevalence ranking No.2 in South Korea. Approximately 20% of the patients are found to be metastatic at diagnosis. It has been reported that 50-60% of the patients who do not have metastasis during the initial diagnosis experience metastasis to other organs. In such cases, the survival rate is only 20.6%. However, treatment options for third-line treatment and above in metastatic patients are limited. Thus, many patients and doctors have voiced high demands for new treatment options that are effective and less burdening. Fruzaqla is a new treatment option for metastatic colorectal cancer that has emerged after over a decade and can be used regardless of specific biomarker status. Fruzaqla was designed to be effective by selectively inhibiting VEGFR-1, 2, and 3. It also minimizes off-targeted toxicity, thereby avoiding unnecessary targets. It has the mechanistic advantage of high-level drug exposure and continuous target inhibition. The efficacy·effectiveness of Fruzaqla has been demonstrated for adult patients with metastatic colorectal cancer who have previously been treated with a chemotherapy containing fluoropyrimidines, oxaliplatin, and irinotecan plus an anti-VEGF or anti-EGFR agent (for patients with wild-type RAS), and whose disease has progressed or who does not show tolerability following treatment with trifluridine/tipiracil and/or regorafenib. The basis of approval was the Phase 3 FRESCO-2 clinical study. The study results showed that the Fruzaqla group had a median overall survival (mOS) of 7.4 months, which was higher than the 4.8 months mOS of the placebo group, and also had a 34% lower mortality risk. Additionally, the Fruzaqla group had a median progression-free survival (mPFS) of 3.7 months, more than double the 1.8 months in the placebo group, corresponding to a 68% reduction in disease progression or death risk. Furthermore, Fruzaqla is an oral treatment that can be taken once daily with convenience without complicated mean conditions. It is expected to yield a positive impact on improving quality of life in addition to the treatment effects. Dr. Sang Cheul Oh, Korea University Guro Hospital (Korean Cancer Study Group's Colorectal Cancer Division Head), said, "Metastatic colorectal cancer, despite its high prevalence and aggressiveness, had been the key cancer type with unmet needs due to limited treatment options for fourth-line or later treatments." Dr. Oh added. "Fruzaqla works by selectively inhibiting VEGFR-1, 2, and 3, and is highly effective but reduced toxicity. It will be a significantly meaningful option for patients at later cancer stages who are undergoing fourth-line or later treatments." Kim Mi-seung, Takeda Pharmaceutical's oncology business unit, said, "Fruzaqla is an innovative new drug that can be used regardless of the specific biomarker status, emerged to the metastatic colorectal cancer treatment setting after 10 years, based on the FDA record. It is expected to solve unmet needs for a wide range of patients," and added," Takeda Pharmaceutical will continue to put efforts in providing improved treatment options for Korean patients, including those for metastatic colorectal cancer."
- Company
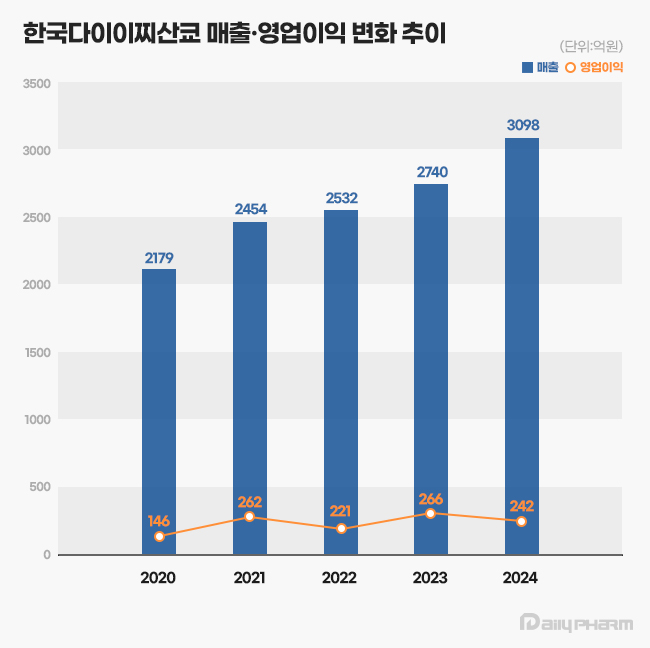
- Daiichi Sankyo exceeds ₩300B in sales…new drug drives
- by Son, Hyung Min Jun 18, 2025 06:01am
- Daiichi Sankyo Korea has exceeded KRW 300 billion in sales for the first time, led by its cardiovascular products, Antibody-Drug Conjugate (ADC), and new anticancer drugs. The company is successfully transitioning its portfolio towards new ADC drugs while maintaining robust growth from established cardiovascular products like Sevikar, Lixiana, and Olmetec. According to the Korea Financial Supervisory Service (FSS)'s electronic disclosure system on June 16, Daiichi Sankyo Korea's sales last year reached KRW 309.8 billion, a 13% increase from the previous year. During the same period, operating profit decreased by 9%, from KRW 26.6 billion to KRW 24.2 billion. Daiichi Sankyo Korea considers its sales for 2024 based on the Japanese fiscal year, covering April of last year to March of this year. Daiichi Sankyo Korea Daiichi Sankyo Korea's sales have been steadily increasing since 2020. The company first surpassed KRW 200 billion in revenue in 2020 with KRW 217.9 billion, followed by a continuous upward trend, reaching KRW 245.4 billion in 2021, KRW 253.2 billion in 2022, and KRW 274.0 billion in 2023. Notably, an analysis suggests that collaboration with the domestic pharmaceutical company Daewoong Pharmaceutical on some cardiovascular products, such as Lixiana and Sevikar, has created a synergistic effect. Daiichi Sankyo Korea signed co-promotion agreements for Sevikar in 2013 and Lixiana in 2015 with Daewoong Pharmaceutical, and this partnership continues to date. Among them, the highest revenue generator is the Direct Oral Anticoagulant (DOAC), Lixiana. According to market research firm UBIST, Lixiana's prescription sales last year was KRW 117.5 billion, a 12% increase compared to KRW 105.3 billion in 2023. DOACs are anticoagulants that prevent blood clots by directly acting on blood coagulation factors. They are increasingly being used in clinical settings as they replace warfarin, which inhibits Vitamin K metabolism. In Korea, Xarelto was approved in 2009, followed by Pradaxa and Eliquis in 2011, and Lixiana in 2015. Despite being the last to be launched among DOACs, Lixiana has rapidly increased its prescription performance, backed by demonstrated clinical data, and has maintained its market dominance since 2019. With annual growth of around 10%, its prescription performance nearly doubled in five years, from KRW 60.4 billion in 2019. Its market share in the overall DOAC market also expanded from 33% in 2019 to 45% last year. Sevikar, an olmesartan-based combination therapy for hypertension, continues to maintain its strong performance in prescription revenue. Sevikar's prescription revenue last year was KRW 68.8 billion, a 4% increase from the previous year. Despite numerous global and domestic pharmaceutical companies entering this market, Sevikar's prescription sales continue to grow. Sevikar's prescription revenue, which was KRW 53.4 billion in 2019, surpassed KRW 60 billion in 2022. In 2023, it recorded KRW 65.9 billion, demonstrating five consecutive years of increased prescription sales. The triple combination hypertension drug Sevikar HCT also maintained its growth trajectory. Sevikar HCT's prescription sales for the last year totaled KRW 42.1 billion, representing a 4% increase from the previous year. Daiichi Sankyo Korea generated approximately KRW 140 billion in prescription sales solely from olmesartan-based hypertension treatments, including Sevikar HCT (KRW 42.1 billion), Olmetec (KRW 30.6 billion), and Sevikar (KRW 68.8 billion). The 5 ADC Strategy...Will it achieve R&D success after Enhertu? Daiichi Sankyo Korea is working towards transitioning from a cardiovascular-focused company to a leader in oncology. The company is particularly concentrating its R&D capabilities on the ADC field, focusing on new growth engines. Following the already approved Enhertu, it is pursuing a '5 ADC strategy' and preparing for the launch of various other therapeutic agents, including Datroway, patritumab deruxtecan, DS-7300, DS-700, and DS-6000. An ADC is a new anticancer drug designed by linking an antibody that binds to a specific target antigen on the surface of cancer cells with a drug that has cell-killing capabilities using a linker. The advantage of ADCs is their ability to selectively target cancer cells by utilizing the antibody's target specificity and the drug's cytotoxic activity, thereby maximizing therapeutic efficacy while minimizing side effects. ADC anticancer agentWhile first-generation ADCs, such as Roche's Kadcyla, were initially limited to breast cancer indications, second-generation ADCs are successfully securing various other indications. Among these, Enhertu is a second-generation new ADC drug introduced by Daiichi Sankyo Korea. Enhertu is a next-generation ADC that links a monoclonal antibody with the same structure as trastuzumab (which binds to specific target receptors overexpressed on the surfaces of cancer cells) and a highly potent, novel topoisomerase I inhibitor payload via a tumor-selective, cleavable linker. Currently, Enhertu won domestic approval for HER2-positive breast cancer, gastric cancer, and non-small cell lung cancer, and is primarily used as a second-line treatment. Its potential as a first-line treatment for breast cancer is also currently being investigated. Daiichi Sankyo is also preparing to launch its second new ADC drug, Datroway. This ADC targets the Trop-2 protein and has been approved in the U.S. for the treatment of breast cancer. The Trop-2 protein is a cell membrane antigen overexpressed in breast cancer, particularly in over 90% of triple-negative breast cancer cases. Datroway binds to the Trop-2 protein and delivers cytotoxic substances into the cancer cells. It has the advantage of maximizing the benefits of targeted therapy and cytotoxic chemotherapy while minimizing damage to healthy cells. Currently, Daiichi Sankyo is co-developing and co-marketing Enhertu and Datroway with AstraZeneca. Daiichi Sankyo is also developing an ADC with Merck. patritumab deruxtecan, which targets HER3, showed efficacy in EGFR-mutated patients compared to platinum-based chemotherapy in the Phase 2 HERTHENA-Lung01 study. Daiichi Sankyo continues to conduct research for subsequent ADC candidates after Enhertu, which targets the HER2 biomarker. The company is also jointly conducting clinical studies with Merck on DS-7300, which targets B7-H3 (an emerging new biomarker in solid tumors), and DS-6000, a CDH6-targeting ADC.

- Company
- Zejula, a new standard ovarian cancer maintenance therapy
- by Whang, byung-woo Jun 18, 2025 06:00am
- Ovarian cancer is often diagnosed at an advanced stage due to the difficulty of early detection, and it is known for its high recurrence rate even after initial treatment. First-line maintenance therapy aimed at delaying recurrence as much as possible after surgery and chemotherapy became a key strategy that determines treatment outcomes for ovarian cancer. Recently introduced PARP inhibitors have emerged as a standard option for maintenance therapy, and the use of biomarkers to guide patient selection has been a major advancement, enabling better prediction of which patient subgroups are likely to benefit the most. In an interview with Dailypharm, Professor Jae Kwan Lee of the Department of Obstetrics and Gynecology at Korea University Guro Hospital, and Dr. Bradley Monk of the Florida Cancer Specialists & Research Institute stressed the need for institutional support for personalized treatment of ovarian cancer. Long-term efficacy of Zejula as first-line maintenance therapy for ovarian cancer proven Ovarian cancer is difficult to diagnose at an early stage and often recurs after initial treatment, raising the importance of maintenance therapy. This is why first-line maintenance therapy to delay recurrence as much as possible after surgery and chemotherapy has become a key strategy in ovarian cancer treatment. Professor Lee said, "First-line maintenance therapy is becoming a critical turning point in ovarian cancer treatment. HRd (homologous recombination deficiency)-positive patients showed an average progression-free survival period extension of approximately 2 years when receiving first-line maintenance therapy after surgery. Given the high recurrence rate of ovarian cancer, maintaining remission for as long as possible is key to successful outcomes, and first-line maintenance therapy serves as a highly effective strategy in this regard." Jae Kwan Lee, Professor of Obstetrics and Gynecology, Korea University Guro Hospital (President, Korean Society of Gynecologic Oncology)One of the changes in the domestic treatment environment for ovarian cancer came with the expansion of reimbursement criteria for the PARP inhibitor Zejula (niraparib) to HRd-positive ovarian cancer in October last year. The reimbursement extension of the PARP inhibitor Zejula was significant because of its biomarker. Approximately 50% of all ovarian cancer patients are HRd-positive, and about half of them, or 25%, have BRCA gene mutations. In addition, studies continue to demonstrate the efficacy of PARP inhibitors in HRd-positive patients. Professor Lee said, “In the past, reimbursement was limited to patients with BRCA mutations, so HRd-positive patients who were BRCA-negative could not choose to use Zejula due to the financial burden. However, since the reimbursement criteria were extended to include HRd-positive patients, many patients are actively starting Zejula treatment.” Zejula is currently one of the most promising PARP inhibitors for first-line maintenance therapy for ovarian cancer. In particular, the long-term follow-up data from the PRIMA study published last year has enhanced the reliability of Zejula. In the PRIMA trial, Zejula increased progression-free survival (PFS) by more than twofold in HRd-positive ovarian cancer patients compared to placebo. Additionally, at the time of clinical confirmation, the median PFS in the Zejula treatment group was 24.5 months, compared to 11.2 months in the placebo group, showing a significant difference. The 5-year PFS rate was also 35%, approximately twice as high as that of the placebo group. Dr. Monk stated, “Previously, there were concerns that long-term use of PARP inhibitors could lead to drug resistance, but this data confirms that such a possibility is low. These long-term follow-up results will serve as a strong source of reliability for doctors who have been hesitant about prescribing Zejula in the long term." He further explained, “Zejula can be used as a first-line maintenance therapy for all patients who respond to platinum-based chemotherapy (all-comer), but it is known to show the most effective results in HRd-positive patients. Since approximately half of all ovarian cancer cases are classified as HRd-positive, Zejula is increasingly being considered as a key option when setting treatment strategies.” “Diagnostic hurdles remain despite Zejula’s extended reimbursement for HRd-positive ovarian cancer” One of the main reasons for the popularity of Zejula is its ease of administration. While other PARP inhibitors require twice-daily dosing, Zejula can be taken once daily, improving medication adherence. Professor Lee said, “For patients to adhere to long-term maintenance therapy without becoming fatigued, treatment convenience is crucial. Zejula’s once-daily dosing regimen has had a positive impact on patients' ability to remain on therapy over the long term without discontinuation." Bradley Monk, MD, Medical Director of Late-Phase Clinical Research Program, Florida Cancer Specialists & Research Institute Dr. Monk added, "Zejula has the advantage of having relatively low drug-drug interactions, which makes it a safer option when used in combination with other drugs. This is a significant advantage for elderly patients with comorbidities or those receiving complex medication regimens.” Meanwhile, with the expansion of reimbursement criteria for HRd-positive ovarian cancer, it has become essential to determine whether a patient is HRd-positive before establishing a treatment strategy, but access to such HRd diagnostic tests remains a barrier. Currently, BRCA1/2 mutation testing for ovarian cancer patients is relatively affordable through national support programs and partial health insurance coverage. However, genomic panel testing required to confirm HRd status is not covered by insurance, leaving patients to bear the full cost of approximately KRW 2.5 million. Professor Lee pointed out, “HRd testing is essential for HRd-positive patients to receive Zejula treatment, but the fact that the test is not covered by insurance and must be paid out of pocket is a major institutional contradiction. Policy improvements should be made so that HRd diagnostic tests can settle as a diagnostic tool accessible under the same criteria as BRCA tests.” In contrast, access to HRd tests has been rapidly improving overseas. Dr. Monk stated, “Currently, more than 10 companies in the United States offer HRd tests, and some provide the service at very low costs. HRd diagnostic tests can serve as an important basis for predicting treatment response to PARP inhibitors such as Zejula.” For this reason, the Korean Society of Gynecologic Oncology is also known to be actively collecting supporting data to officially propose reimbursement for HRd tests to Korean health authorities. If HRd tests are promptly covered by health insurance, patients will be able to receive the necessary testing without financial burden and fully enjoy the benefits of targeted maintenance therapy such as Zejula. In addition, Professor Lee, who has been appointed as the President of the Korean Society of Gynecologic Oncology, emphasized his commitment to advancing precision medicine based on the genetic profiling of ovarian cancer. Professor Lee stated, “The society plans to focus on how to diagnose and manage the genetic characteristics of ovarian cancer. In other countries, there are already detailed clinical guidelines in place for individuals with BRCA mutations, and I believe similar protocols are needed in Korea as well." He concluded, "Since ovarian cancer often occurs alongside other cancers such as breast or endometrial cancer, collaboration with other specialties, including surgical departments, is essential. Establishing a multidisciplinary, patient-centered integrated care system through close coordination with various medical fields is another key priority for the society."
- Company
- Adstiladrin receives orphan drug designation in Korea
- by Eo, Yun-Ho Jun 18, 2025 05:59am
- The new bladder cancer drug Adstiladrin has been designated as an orphan drug in Korea. The Ministry of Food and Drug Safety recently announced the news in a orphan drug designation announcement. The specific indication for designation is “treatment of BCG-refractory high-risk non-muscle-invasive bladder cancer (NMIBC) with carcinoma in situ (CIS) with or without papilloma.” Adstiladrin (nadofaragene firadenovec-vncg) received FDA approval in the United States in 2022. This drug uses a non-replicating adenovirus vector to deliver the human interferon alpha-2b gene, inducing an immune response by directly expressing the protein within the bladder epithelium. Adstiladrin demonstrated efficacy through the NCT02773849 clinical trial that involved 157 patients with bladder cancer. In the study, 51% of the 98 patients treated with Adstiladrin achieved complete response (CR). The median duration of response was 9.7 months. In addition, 46% of patients who achieved CR remained recurrence-free at 12 months after treatment. The most commonly reported side effects were instillation site discharge (33%), fatigue (24%), bladder spasms (20%), urinary urgency (19%), and hematuria (17%). The rate of discontinuation due to side effects was 1.9%. Non-muscle-invasive bladder cancer (NMIBC) is an early-stage bladder cancer confined to the bladder mucosa, accounting for approximately 70–80% of all bladder cancers. Among these, high-risk patients include those with carcinoma in situ (CIS) or multifocal high-grade tumors, which have a high risk of recurrence and invasion. Although BCG instillation therapy is used as first-line treatment, approximately 30–50% of patients eventually experience recurrence or become resistant within a few months. While radical cystectomy is considered the standard treatment thereafter, as it is a highly invasive surgery, there has been a continued demand for bladder-preserving therapeutic alternatives.