- LOGIN
- MemberShip
- 2026-05-03 09:21:14

- Company
- Is the heyday of domestic flu vaccines coming to an end?
- by Moon, sung-ho Sep 10, 2024 05:48am
- Competition among pharmaceutical and biotech companies with influenza (flu) vaccines is intensifying more than ever. As more and more companies, including global pharmaceutical companies, are seeking to supply vaccines on-site, and specialized vaccines are being launched for each age group, the competition is expected to be unprecedented this autumn. # In particular, the formation of a non-reimbursed market for both vaccines has become a hot topic in the field, especially in the context of the resurgence of COVID-19. However, industry insiders do not expect large-scale co-vaccination to be carried out among adults, except for high-risk patients who are eligible for the National Immunization Program (NIP). According to industry sources on the 7th, the vaccine contracted by the Korea Disease Control and Prevention Agency (KDCA) for the ‘2024-2025 Influenza NIP Project’ is 11.7 million doses worth KRW 12.9 billion. This is more than the previous year's contracted volume of 11.21 million doses. KDCA signed contracts with GC Biopharma, Sanofi, Boryung Biopharma, SK Bioscience, Korea Vaccine, and Il-Yang Pharmaceutical. This is similar to last year. However, the details are different. Last year, GC Biopharma supplied less than planned after making the highest bid through the NIP, but the company made a different choice this time. Last year, GC Biopharma bid to supply 4.3 million doses, but only 1.74 million doses were delivered through NIP due to its supply price being pushed back. In other words, the doses that the company was unable to supply through the NIP were released into the non-reimbursed adult vaccine market. But the situation was different this year. GC BIopharma bid the highest price for the NIP, at KRW 10,810 per vaccine, and the largest volume of 2.65 million doses. The company was followed by SK Bioscience, which bid KRW 10,470 per vaccine for 2.55 million doses. It is also worth noting that Sanofi, which bid the lowest price at KRW 10,340, will supply 2.15 million doses. In addition, Il-Yang Pharmaceutical and Korea Vaccine will supply 2 million doses and Boryung Biopharma will supply 1.25 million doses to NIP. In addition, GSK will supply the flu vaccine Fluarix Tetra for the pediatric NIP. As a result, most of the flu vaccines are included in the NIP. The difference this year is in the adult vaccine market for the elderly. This is because Sanofi is planning to launch ‘Efluelda,’ a high-dose influenza vaccine for people aged 65 and over in time for this year's flu season in addition to the existing Vaxigrip. It is worth noting that CSL Seqirus launched ‘Fluad Quad’ last year for the 65+ age group. Fluad Quad was the first vaccine to enter this market, but the subsequent entry of Efluelda has created competition in the non-reimbursement vaccine market for the 65+ age group. While the NIP includes people aged 65 and older, clinical sites say there is a clear market for non-reimbursed Fluad Quad given the vaccination experience last year. “Given the situation last year, 1 in 10 of the elderly sought the vaccine,” said an otolaryngologist who requested anonymity. “I brought in about 100 units, which I used with the 1,200 doses of the NIP vaccine. I think the point that it is particularly effective for those aged 65 and older has made them aware of the need.” “Last year, the price of the vaccine was KRW 40,000 to 50,000, but this year, we need to consider the entry of its competitor,” he said. “There may be differences by region, but the price of the general vaccine is likely to be between KRW 25,000 and 30,000, and the price of the vaccine for the elderly is likely to set KRW 10,000 higher.’ Another point of interest is how the reemergence of COVID-19 will affect the flu vaccination season later this year. According to KDCA, 837 hospitalized patients were reported in the COVID-19 sentinel surveillance report at Week 35, a 28.0% decrease from Week 34 (1,163). This is a 48.2% decrease compared to Week 33 (1,464), 2 weeks earlier. The pathogen detection rate has also been decreasing for 2 consecutive weeks, from Week 33 (43.4%) to Week 34 (39.0%) to Week 35 (34.0%). COVID-19 viral concentrations in the wastewater surveillance also decreased by 27% from 2 weeks ago. While it is believed that the peak of the pandemic has passed, the government plans to continue to support COVID-19 vaccinations for high-risk groups, including those aged 65 and older and those who are immunocompromised. The clinical field is interested in the volume of non-reimbursed COVID-19 vaccinations alongside the flu vaccinations. If interest continues to rise, the patients may inquire about COVID-19 vaccinations during the flu vaccination season. In this situation, Moderna Korea, a leading vaccine supplier, has ended its relationship with Kwangdong Pharmaceutical and entered into a strategic partnership with Boryung Biopharma for the supply of COVID-19 vaccines in Korea. Through the partnership, Boryung Biopharma will provide medical information on Moderna's updated COVID-19 vaccine, which will be used in the government's ‘2024-2025 Season Inoculation Program,’ to healthcare providers in Korea. However, it is unlikely that many adults will be vaccinated with the non-reimbursed COVID-19 vaccine as it is relatively expensive. Currently, there is a consensus that the vaccine price will be in the mid-KRW 100,000 range without reimbursement. “The government seems to want to support COVID-19 vaccines to high-risk groups such as the elderly over the age of 65. Some of the other COVID-19 vaccines are currently in supply and their purchase price is in the KRW 100,000 range,” said Kyung-Geun Kwak, Chair of Seoul Physician’s Association (Seoul Medical Clinic) ”Because of this, the price of the COVID-19 vaccination will be in the mid- KRW 100,000 range. The flu had spread in Korea from August to September along with COVID-19, so those who had suffered inconvenience due to the 2 may likely seek vaccination for either disease.” Kwak added, “I understand that there are global policies in place, but personally I believe the COVID-19 vaccine is too expensive. “I doubt that relatively younger patients, except for high-risk groups, would be willing to receive vaccination at the price.”

- Company
- Livtencity's introduction raises expectations in the field
- by Hwang, Byung-woo Sep 10, 2024 05:47am
- The introduction of Livtencity (maribavir), a drug that can be prescribed to manage infections in transplant patients following the use of existing treatments, has been met with positive reviews in the field due to a lack of treatment options. Although the actual number of patients who will be prescribed the drug is limited, the drug is expected to play a role in the management of infections in patients who value their transplant opportunity. Sung-Han Kim, Department of Infectious Diseases, Seoul Asan Medical Center Sung-Han Kim, Professor of Infectious Diseases at Seoul Asan Medical Center, attended a media session organized by Takeda Pharmaceuticals Korea to discuss the paradigm shift in the treatment of patients with cytomegalovirus (CMV) infection. CMV is a double-stranded DNA virus that is a member of the herpesvirus family and is mainly transmitted through body fluids, white blood cells, and tissues such as transplanted organs. In Korea, about 94% of adults are known to be seropositive for CMV. For this reason, allogeneic hematopoietic stem cell transplantation or solid organ transplantation patients, whose immune function is temporarily reduced to control the body's rejection during the transplantation process, may develop latent CMV, rendering it an essential condition to manage. In Korea, up to 88% of patients after allogeneic stem cell transplantation and up to 55% of patients after solid organ transplantation experience CMV infection. CMV infection may be initially asymptomatic, but if not successfully treated, it can progress to CMV disease. CMV infection is also considered a major threat that increases the risk of graft rejection, opportunistic infections, and death. “People can be infected with CMV during adolescence to about 25 years of age and remain latent,” said Professor Kim, “and it can cause a number of diseases when the immune system is suppressed following transplantation. CMV infection in transplant patients is an indication of a poorer immune status, so it needs to be managed appropriately.” Currently, CMV treatment is divided into three phases: prophylaxis for those who do not have the virus, preemptive therapy for those who do have the virus but have not developed a disease, and treatment for those who have advanced to a disease. For solid organ transplant and allogeneic stem cell transplant patients, who are at high risk for CMV, ganciclovir (intravenous) and valganciclovir (oral) are used as first-line treatments. In the case of patients who show resistance or are refractory to antivirals and require second-line treatment, solid organ transplant patients are treated with ganciclovir or valganciclovir in combination with an adjusted dose of immunosuppressive agents. Foscarnet and cidofovir may also be considered, but their use is limited due to their non-reimbursed status. ”Livtencity is expected to serve as a promising new weapon for treatment of CMV...changing the guideline protocols remains a challenge” Livtencity has emerged in this situation. The drug was launched last year and has been granted reimbursement as a second-line treatment since April. Although the number of transplant patients is limited in Korea, the significance of its arrival is that it brings additional options to the field. Although several drugs are already used for CMV, there are patients who are refractory to the existing options, so Livtencity is expected to play a role as a later-line option. Kim said, “Organ transplants and bone marrow transplants are expensive, and there are cases where CMV can ruin the transplant. In an area where options are limited, Livtencity’s introduction is significant because it has a better effect and fewer side effects.” ‘As Livtencity is an oral pill, it can be taken as an outpatient treatment, which makes it more convenient for the patients,” added Kim. However, Kim believes that guidelines for post-transplant infection control need to be contemplated. While the growing options have improved patient access, it also comes with the risk of cuts. For allogeneic hematopoietic stem cell transplantation, the current treatment sequence is to use conventional first-line treatments such as ganciclovir and valganciclovir followed by Livtencity as a second-line treatment. MSD's Prevymis (letermovir) has also emerged as a prophylaxis for CMV infection, but it can only be used for up to 100 days after transplantation. “Prophylaxis after allogeneic stem cell transplantation is done most of the time, but due to the high cost of the drug and concerns about reimbursement cuts, the treatment is sometimes terminated at the wrong time or the right dose not used,” said Kim. “There is also a need for the hematology and infectious disease departments to discuss the treatment protocols to manage CMV after allogeneic stem cell transplantation.” “Various discussions need to be made to address these protocol issues, which is not easy in the current environment. We would need to sort out these protocols after the overall healthcare situation improves in Korea.”

- Company
- Eliquis generics to enter and reshape DOAC mkt in SEP
- by Moon, Sung-ho Sep 10, 2024 05:47am
- From this month (September), numerous generic drugs will be released in the direct-acting oral anticoagulant (DOAC) market. Following Bayer's Xarelto (rivaroxaban), Pfizer and BMS's Eliquis (apixaban) is also facing its patent expiry. According to industry sources on the 26th, the Ministry of Health and Welfare recently prepared a notice on the reimbursement listing of 18 pharmaceutical companies' products containing apixaban and began collecting opinions. When looking at the DOACs that are prescribed in internal medicine hospitals and clinics, Bayer’s Xarelto (rivaroxaban), followed by Boehringer Ingelheim’s Pradaxa (dabigatran), Pfizer and BMS’s Eliquis (apixaban), and Daiichi Sankyo’s Lixiana (edoxaban) are currently the major players in Korea’s market. Among these, Xarelto’s patent expired in the second half of 2022, and its decline in the prescription market has become more pronounced recently as domestic drugmakers have launched a slew of Xarelto generics. According to the drug research institution UBIST, Xarelto’s prescriptions plummeted 37% from KRW 49.4 billion in 2022 to KRW 31 billion in 2023. The downward trend has continued in the first half of this year, with sales around KRW 15.3 billion. Moreover, as another blockbuster, Eliquis, is scheduled to expire on September 10, the MOHW has announced that 18 Eliquis generics from pharmaceutical companies will be listed for health insurance reimbursement benefits. For reference, Eliquis continues to dominate the clinical scene with prescriptions worth KRW 77.3 billion last year. In the first half of this year, it recorded KRW 38.8 billion, but this sales flow is expected to change after the launch of generics. Upon the entry of generics, Daiichi Sankyo's Lixiana, which has recently been strengthening its market dominance, is expected to take the sole lead in the field. Lixiana, which is copromoted and sold by Daiichi Sankyo and Daewoong Pharmaceutical, recently surpassed Eliquis’s sales and rose to monopoly. Last year, Lixiana's domestic sales reached KRW 105.3 billion, and in the first half of this year, it recorded KRW 55.8 billion, maintaining an upward trend. A professor of cardiology at A University Hospital said, “Nothing would notably change in the field aDOACs are well used in clinical practice. If generics are released, prescriptions will naturally be dispersed between them as they may differ in terms of drug price.” A director of a frontline internal medicine clinic also said, “Following Xarelto, the patent expiry of major original drugs in the DOAC market has followed. I think Eliquis’s market will show a similar result to Xarelto’s. We have already been approached by sales representatives from various pharmaceutical companies requesting prescriptions from September following the launch of their respective generics.” “I can feel the pressure put on the sales representatives by pharmaceutical companies that have launched generics. Everybody rep had been asking us to just prescribe one case,” added the director.

- Company
- Hugel hosts Train-The-Trainer seminar for global medical aes
- by Kwon Sung-Yong Sep 09, 2024 09:45am
- Hugel Inc., a leading global medical aesthetics company, said on Monday it hosted the Train-The-Trainer program at the end of August with medical experts from overseas to improve their knowledge and practices of skin rejuvenation. Some 20 medical practitioners from nine countries, including the US, Australia, Canada, China, Taiwan, Japan, Indonesia, Kazakhstan and Colombia, attended the two-day seminar in Seoul with lectures and cadaveric demonstrations. The company hosted a cadaveric workshop at the Catholic Institute for Applied Anatomy of the Catholic University of Korea on the first day. The injection of botulinum toxin and hyaluronic acid fillers, which helps facial rejuvenation and a youthful gaze, and theory courses were led by Dr. Kyuho Yi, the director at Maylin Clinic Apgujeong, Soo-Bin Kim, professor at the Department of Anatomy of Yonsei University College of Dentistry and Hyun Jin Park, professor at Department of Anatomy of Daegu Catholic University School of Medicine. On the second day, the seminar was focused on advanced aesthetic injection techniques. Lectures and live demonstrations for skin rejuvenation were performed by Dr. Ho Sung Choi, the medical director of Piena Aesthetic Clinic, and Dr. Jong Jin Lee, the medical director of DayBeau Clinic. “The trainees were highly satisfied with the seminar which facilitated sharing injection techniques of Korean medical experts, by using Hugel products, deep knowledge and emerging trends in the medical aesthetics field via a well-structured curriculum. The training course helps experts from different markets grow as global medical aesthetic industry leaders. Hugel will continue to actively organize academic events in each market to improve medical practices for skin rejuvenation,” a Hugel official said. Hugel hosted Train-The-Trainer for the second time after the first one held in April in Seoul. The course in August was part of Hugel Expert Leader’s Forum (H.E.L.F.), the global academic forum for medical aesthetic professionals that the company has held every year since 2013. About Hugel Established in 2001, Hugel is a leading global medical aesthetics company that manufactures injectables for skin rejuvenation such as botulinum toxin, hyaluronic acid fillers and skin boosters as well as tissue-lifting threads and cosmetics products. The company is the only South Korean supplier to the world’s three largest botulinum toxin markets, the US, China and Europe. It exports medical aesthetic products to around 70 countries and operates eight global subsidiaries in the US, Australia, Canada, Taiwan, China, Hong Kong and Singapore.
- Company
- 'Vabysmo' likely to receive approval for third indication
- by Eo, Yun-Ho Sep 09, 2024 05:49am
- Product photo of Roche Korea 'Vabysmo,' the first bispecific antibody for the treatment of ophthalmologic disease, is under review for expanded indication for retinal vein occlusion (RVO) in South Korea. According to industry sources, Roche Korea has applied for expanded approval of Vabysmo (faricimab) from the Ministry of Food and Drug Safety (MFDS). The drug received approval for the RVO indication from the U.S. Food and Drug Administration (FDA) last October. Vabysmo is a treatment for macular degeneration that draws attention for significantly extending the administration interval compared to 'Eylea (aflibercept),' which has been the standard therapy. In South Korea, Vabysmo's prescription became available after it was approved for reimbursement listing for neovascular age-related macular degeneration (nAMD) and diabetes-related macular edema (DME) in October last year. Existing macular degeneration drugs used in South Korea are vascular endothelial growth factor-A (VEGF-A) drugs such as Novartis' 'Lucentis (ranibizumab),' 'Beovu (brolucizumab),' and Eylea. Unlike existing VEGF drugs, like Lucentis and Eylea, Vabysmo can also block the angiopoietin-2 (Ang-2) pathway, thus inhibiting new blood vessel formation. The analysis suggests that blocking two independent pathways can more effectively stabilize blood vessels and reduce inflammation, abnormal vessel growth, and fluid leakage than the VEGF-A pathway alone. RVO is Vabysmo's third indication. Its efficacy has been confirmed through the Phase 3 BALATON and COMOINO studies. In these clinical trials, Vabysmo achieved non-inferiority in the patient's vision improvement compared to Eylea. When treated with Eylea, the patients had continual vision improvements from the early stage. The safety profile of the trials was similar to previous study reports. According to the market research firm IQVIA, the domestic market for macular degeneration treatment generates KRW 110 billion in sales as of 2021. Eylea accounts for KRW 70.5 billion, and Lucentis accounts for KRW 35.1 billion of the total sales. The future impact of Vabysmo, which is expanding into various fields, on the market is to watch.

- Company
- Jassen’s Balversa can be prescribed in general hospitals
- by Eo, Yun-Ho Sep 09, 2024 05:49am
- The new bladder cancer drug Balversa may now be prescribed in general hospitals in Korea. According to industry sources, Janssen Korea’s FGFR-inhibiting urothelial carcinoma (bladder cancer) drug Balversa (erdafitinib) has recently passed the drug committees (DCs) of tertiary hospitals including Seoul St. Mary's Hospital, Seoul Asan Medical Center, and Sinchon Severance Hospital, as well as the drug committees of medical institutions such as Hallym University Kangnam Sacred Heart Hospital, National Cancer Center, and Cheonam National University Hwasun Hospital. Balversa was approved by the Ministry of Food and Drug Safety in January 2022. However, it is still not reimbursed in Korea. Specifically, the drug is indicated for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma (mUC) with FGFR2 or FGFR3 genetic alterations whose disease has progressed on or after at least one line of prior systemic therapy, which includes platinum-based chemotherapy, or whose disease has progressed within 12 months of neoadjuvant or adjuvant treatment with platinum-based chemotherapy. However, the approval of PD-1 and PD-L1-directed immuno-oncology agents in the first- and second-line settings that followed Balversa’s approval led to the need for Balversa to demonstrate efficacy in patients who previously received these agents. And the situation was addressed with the publication of Balversa’s Phase III THOR trial study, which demonstrated a prolonged overall survival (OS) benefit with Balversa over chemotherapy in patients with metastatic urothelial carcinoma with FGFR3/2 gene alterations whose disease progressed after first-line treatment with immuno-oncology agents. In the study, Balversa prolonged overall survival (OS) compared with chemotherapy in patients with metastatic urothelial carcinoma. Results showed that over a median follow-up of 15.9 months, the mOS was 12.1 months in the Balversa arm, reducing the risk of death by 36% compared with the 7.8 months in the chemotherapy arm. Based on these findings, the U.S. Food and Drug Administration granted Balversa formal approval in January, but with a more restricted indication than originally approved. The European Medicines Agency's Committee for Medicinal Products for Human Use recently recommended expanded indications for Balversa. Janssen Korea has also additional submitted results from the THOR study to Korea’s Ministry of Food and Drug Safety. Therefore, the company well may launch Balversa in earnest in the second half of the year in Korea. It remains to be seen whether Balversa will be able to go beyond landing in medical institutions and gain insurance coverage in Korea. Meanwhile, bladder cancer is one major cancer that has lacked a targeted therapy option. Balversa is the first targeted anti-cancer drug for bladder cancer with a novel mechanism of action that inhibits fibroblast growth factor receptor (FGFR). FGFR is a biomarker involved in cancer cell growth that is associated with various cancers. FGFR mutations are particularly common in bladder cancer, with 20 to 30% of patients carrying mutations.
- Company
- Vonjo receives orphan drug designation in Korea
- by Eo, Yun-Ho Sep 06, 2024 05:48am
- The oral myelofibrosis drug Vonjo has been designated as an orphan drug in Korea. The Ministry of Food and Drug Safety announced so on the 3rd through an orphan drug designation. Specifically, the drug is indicated for ‘adults with intermediate or high-risk primary or secondary (post-polycythemia vera or post-essential thrombocythemia) myelofibrosis (MF) with a platelet count

- Company
- SGLI2i and dulaglutide similar in reducing dementia risk
- by Hwang, Byung-woo Sep 06, 2024 05:48am
- With interest in the dementia prevention effect of Type 2 diabetes medications such as SGLT-2 inhibitors and GLP-1 RAs rising, a study has been published in Korea on the relative prevention effect between the two medications. (from the left) Professor Ju-young Shin, Ph.D candidate Bin Hong, Dr. Sungho Bea, Ph.D candidate Hwa Yeon Ko On the 3rd, Sungkyunkwan University College of Pharmacy Professor Ju-young Shin’s research team (First author: Bin Hong, coauthor Sungho Bea) announced today that it had published results of a research comparing the dementia prevention effects of SGLT2 inhibitors and dulaglutide in patients with type 2 diabetes using Korea’s healthcare big data. Dementia is a debilitating neurodegenerative disease characterized by cognitive decline and memory loss. Type 2 diabetes is an important risk factor for dementia, and people with Type 2 diabetes have a significantly increased risk of developing dementia compared with those without. Given the increasing prevalence of Type 2 diabetes and the burden of dementia, an urgent need exists to find effective strategies to prevent or delay the development of dementia in patients with Type 2 diabetes. In particular, two classes of diabetes medications, SGLT2 inhibitors and glucagon-like peptide-1 receptor agonists (GLP-1 RAs), have attracted much attention in recent years due to their excellent effects on cardiovascular and renal function in addition to their blood glucose lowering effects. Recent studies have also shown that SGLT2 inhibitors and GLP-1 RAs may also have neuroprotective effects. According to the research team, its research comparing the effects of these two drug classes on cognitive function and dementia risk had limitations, as it was a small, randomized clinical trial with a small sample size of 36 participants and relatively short follow-up periods of 16 weeks. In addition, patients with a history of psychiatric disorders, which are associated with a higher risk of developing dementia, were excluded, so the results may not accurately reflect the risk of dementia in patients taking this drug in a real-world setting. Therefore, the effects of SGLT2 inhibitors and GLP-1 RAs on cognitive function and dementia risk in patients with Type 2 diabetes remain unclear. Using target trial emulation, Professor Shin’s research found that there was no significant difference in the risk of dementia between SGLT2 inhibitors and dulaglutide (the most commonly used GLP-1 RA in Korea) in patients with Type 2 diabetes in the real-world setting. The research was conducted on patients aged 60 years or older who have type 2 diabetes and are initiating treatment with SGLT2 inhibitors or dulaglutide from May 1, 2016, to December 31, 2020. The final cohort was set by taking into account the patient’s age, gender, diabetes severity, concomitant medications, co-morbidities, medical examination results, and risk factors for dementia based on his or her past year’s medical history to calculate a propensity score. Within the propensity score-matched cohort, 2,076 patients prescribed SGLT2 inhibitors and 1,038 patients prescribed dulaglutide were included in the research. During a median follow-up of 4.4 years, 69 patients in the SGLT2 inhibitor group and 43 patients in the dulaglutide group developed dementia. Comparing the SGLT2 inhibitor group to the dulaglutide group, the five-year difference in dementia risk was -0.91 percentage points (95% CI, -2.45 to 0.63 percentage points), with a hazard ratio of 0.81 (CI, 0.56 to 1.16). “Given the lack of conclusive evidence to support specific drug treatments aimed at preventing dementia in current guidelines, this study is significant in that it generates evidence by directly comparing the risk of dementia for 2 novel diabetes medications - SGLT2 inhibitors and GLP-1 RAs,” said Professor Shin. “We found no significant difference in the risk of dementia between SGLT2 inhibitors and dulaglutide, but it is uncertain whether these results can be generalized to newer GLP-1 RAs. Thus, further studies incorporating newer drugs within these drug classes and better addressing residual confounding are required,’ the researchers concluded. The study was conducted by Professor Shin’s research team at Sungkyunkwan University in collaboration with Professor Woo Jung Kim, Department of Psychiatry at Yongin Severance Hospital, and Professor Young Min Cho, Department of Endocrinology at Seoul National University Hospital. The study used customized data from the National Health Insurance Service and was supported by the Ministry of Food and Drug Safety. The results were published online on August 27 in the Annals of Internal Medicine (IF: 19.6, JCR Ranking 2.3%), a prestigious international journal in the field of medicine.

- Company
- K-made new anti-cancer drugs to unveil at conference
- by Son, Hyung-Min Sep 06, 2024 05:48am
- South Korea-based pharmaceutical and biotech companies will unveil their clinical results at the European Society for Medical Oncology (ESMO) Congress (ESMO 2024), which will be held for four days from September 13th. Korean pharmaceutical and biotech industry will unveil their positive clinical trial results at the world's largest cancer conference. The clinical outcomes of various new drug candidates from the South Korea-based pharmaceutical and biotech companies will be presented at the European Society for Medical Oncology (ESMO) Congress (ESMO 2024), which will be held for four days from September 13th. ESMO is one of the three renowned cancer associations, next to the American Association for Cancer Research (AACR) and the American Society of Clinical Oncology (ASCO). Rivoceranib demonstrated effectiveness in various solid cancers HLB group will unveil research outcomes for its targeted therapy Rivoceranib for various cancers, including liver cancer, cholangiocarcinoma, esophageal cancer, melanoma, thyroid cancer, and ovarian cancer. According to industry sources on September 5th, during ESMO 2024, HLB group will unveil research outcomes for its targeted therapy Rivoceranib for various cancers, including liver cancer, cholangiocarcinoma, esophageal cancer, melanoma, thyroid cancer, and ovarian cancer. HLB group and Jiangsu Hengrui Pharmaceuticals are evaluating the clinical efficacy of the combination of Rivoceranib, a vascular endothelial growth factor receptor-2 (VEGFR2) inhibitor, and camrelizumab, an immune checkpoint inhibitor. In May, the companies applied for U.S. Food and Drug Administration (FDA) approval for Rivoceranib in combination with camrelizumab, a PD-1 immune checkpoint inhibitor, as a new drug for liver cancer. However, they received a complete response letter (CRL) request. In liver cancer, Rivoceranib plus camrelizumab recorded the final overall survival (OS) of 23.8 months, which was most prolonged result among other competing drugs. During ESMO 2024, the companies will unveil additional analysis on quality-of-life improvement of the combination therapy in patients with liver cancer. HLB group and Jiangsu Hengrui Pharmaceuticals will also unveil the positive results of the combination therapy for various solid cancers, including cholangiocarcinoma and thyroid cancer. The results from the clinical trial involving 28 patients with cholangiocarcinoma showed that patients treated with Rivoceranib plus camrelizumab combination therapy had a median OS of 12.8 months and a progression-free survival (PFS) of 6.3 months. They confirmed twice more prolonged survival than the average 6-7 months for non-operable patients. Patients treated with Rivoceranib plus camrelizumab combination therapy had a 50% objective response rate (ORR). The results of the Rivoceranib monotherapy for thyroid cancer will be presented. According to the clinical results to date, 13 patients with thyroid cancer who have taken Rivoceranib showed an ORR of 53.8% in perioperative adjuvant therapy. Disease control rate (DC) showing cancer that is maintained without reduction or enlargement were observed in all patients. Post-surgical results following Rivoceranib monotherapy showed 84.6% complete resection, where the remaining cancer cells are no longer detected. During ESMO 2024, HLB group will also unveil the final clinical results of the Rivoceranib plus camrelizumab combination therapy used as adjuvant therapy for patients with esophageal cancer. Studies for potential drugs with various mechanisms of actions, immune checkpoint inhibitors·CAR-T, are underway TiumBio, ABION BIO, and Eutilex will present the clinical trial results of new drug candidates with various mechanisms of action, such as immune checkpoint inhibitors and chimeric antigen receptor-T cell (CAR-T) therapies. During the conference, TiumBio will unveil the additional results of the Phase 1b trial for TU2218, an immune checkpoint inhibitor candidate, in combination with MSD's immune checkpoint inhibitor Keytruda. The results to be disclosed will include the safety data of the combination therapy and the anti-tumor responses of patients with advanced solid cancers. TU2218 blocks pathways of transforming growth factor beta (TGF-ß) and vascular endothelial growth factor (VEGF), which are known to hinder cancer immunotherapy activation. TU2218's mechanism maximizes the efficacy of cancer immunotherapy. TiumBio is conducting a Phase1b trial in three clinical institutes in the United States to evaluate the efficacy and safety of TU2218 in combination with Keytruda in patients with advanced solid tumors. During the Phase 1b trial, TiumBio confirmed partial response (PR) from two patients and stable disease (SD) from three patients out of five patients who are evaluable for efficacy. Also, TU2218 showed an ORR of 40% and a disease control rate (DCR) of 100%. ABION BIO will unveil the clinical results of vabametkib, a candidate product for targeted treatment of non-small cell lung cancer (NSCLC). Vabametkib targets NSCLC's c-MET mutations. C-MET is one of the proteins transmitting a signal to cells expressing the MET (MNNG HOS transforming gene) gene. It is considered a cancer-causing gene. This gene is associated with various solid cancers, including lung cancer, colorectal cancer, gastric cancer, and liver cancer. C-MET mutations are known to occur in 6% of all NSCLC patients. Based on the clinical results, treating vabametkib in patients with c-MET mutation NSCLC who failed prior therapy had an ORR of 52.9%. Patients who had not received prior treatment had an ORR of 75%. Eutilex will participate in a poster session, presenting the clinical results of its CAR-T candidate EU307 for patients with liver cancer. EU307 is a CAR-T treatment targeting GPC3, which is abnormally overexpressed in liver cancer cells without affecting healthy cells. EU307 has been designed to raise the CART-T function by inhibiting the immune-related cytokine, 'interleukin (IL)-18,' and to improve the tumor microenvironment (TME). The TME blocks immune cells from entering tissues located near cancers, inhibits cancer metastasis, and improves survival. EU307's clinical trial was designed as Phase 1, a dose-escalation multi-center approach to evaluate the safety, tolerability, pharmacokinetics (PK), pharmacodynamics (PD), and anti-tumor activity. The study participants included 'patients with GPC3-positive advanced liver cancer who failed the standard therapy.'
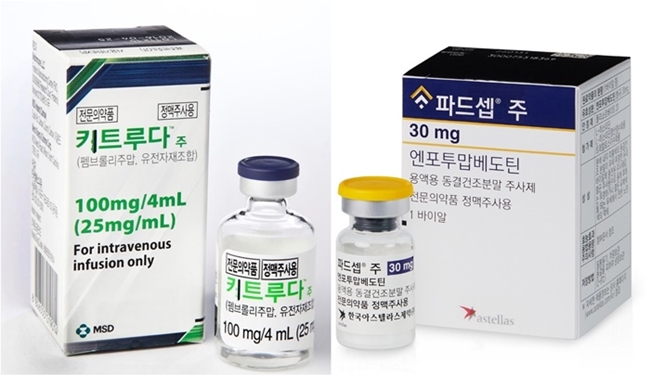
- Company
- Padcev’s reimb complex due to considerations
- by Hwang, Byung-woo Sep 05, 2024 05:52am
- Astellas Pharma Korea is continuing complex calculations for health insurance reimbursement listing of its metastatic urothelial cell carcinoma drug Padcev (enfortumab vedotin) in Korea. The reimbursement is being discussed for the second-line monotherapy indication, which was first approved. However, as the company is seeking reimbursement for the potentially high-impact Keytruda+Padcev combination therapy, more factors remain in need of consideration. (From the left)Keytruda, Padcev According to industry sources, Astellas Pharma Korea is planning to file a reimbursement application for the Keytruda+Padcev combination therapy as a first-line treatment for urothelial cancer later this year. The clinical value of the combination is unquestionable, as it is already raising expectations as a treatment that will change the first-line treatment paradigm of urothelial cancer in 30 years. “Recently, a variety of innovative new drugs have emerged in urothelial cancer, and Padcev is the first new ADC drug for metastatic urothelial cancer that has settled as a new first-line standard of care option in 30 years,” said In-Keun Park, Professor of Medical Oncology at Asan Medical Center in Seoul. “The drug is driving a major shift in treatment strategy, and global guidelines recommend Padcev as the only first-line option for the treatment of urothelial cancer.” The challenge is that this paradigm-shifting option for first-line treatment of metastatic urothelial cancer is a combination therapy. The combination of Astellas' Padcev and MSD's Keytruda requires coordination between the two companies. According to industry sources, Astellas does not need MSD's consent to apply for reimbursement for the Padcev+Keytruda combination, because Astellas is only seeking reimbursement for Padcev part of the combination. This means that Astellas will have to apply for Padcev’s reimbursement in the Padcev+Keytruda combination and wait to see how the situation develops. Currently, there is no clear track for partial reimbursement of combination therapy, and further discussions will depend on whether the Health Insurance Review and Assessment Service decides to consider the benefits of the combination on a case-by-case basis and grant partial coverage, or whether it will consider reimbursing Keytruda as well. “Just as the combination therapy of Padcev+Keytruda was approved and the label was separately updated by each pharmaceutical company, the reimbursement does not need to be discussed between the companies,” said an industry insider. “The reimbursement application contains sensitive information about each company, such as the way each drug’s reimbursement is listed and the drug price, so the process needs to be done separately rather than discussed between the 2 companies.” He added, “While there is a precedent for Imfinzi, it is difficult to predict how the government will approach the Padcev+Keytruda combination.” However, in this case, MSD's willingness to reimburse its part became important because the reimbursement of only half of the combination is impractical. According to industry sources, MSD has also been having internal discussions about applying for reimbursement for the Padcev+Keytruda combination. For Astellas, the extent to which MSD is willing to focus on reimbursement in the future will serve as a variable, especially as there are concurrent reimbursement applications for Keytruda monotherapy in progress other than the one for the Padcev+Keytruda combination. Drug price negotiations and other variables remain for Padcev’s pending second-line reimbursement Another concern for Astellas is the delay in reimbursement discussions for Padcev’s reimbursement in the second line. Padcev passed HIRA’s Cancer Disease Review Committee in February but was unable to cross the threshold of the Drug Reimbursement Evaluation Committee, due to DREC’s request for supplementary data. In addition, even if the drug passes DREC review in the future, it will take more time for the drug to be added to the reimbursement list due to drug price negotiations. As the insurance drug price set for the second line could serve as a reference point when the company seeks to extend the drug’s coverage to first-line treatment, the government and the company may have different views at the drug price negotiation stage. One thing to look forward to is that Padcev qualifies as an innovative new drug in Korea. Recently, HIRA established a new flexible ICER threshold for innovative new drugs. “Since (the combination therapy) is a new type of reimbursement model, we are internally discussing which method will be the fastest and most effective,” said Kyung-ah Park, Director of Medical Affairs at Astellas Korea. “We see it as our responsibility to reimburse Padcev and are actively working on it, and as Padcev meets the three criteria as a Korean innovative new drug, we plan to improve access through reimbursement as soon as possible.