- LOGIN
- MemberShip
- 2026-05-08 01:10:31
- Company
- Promoting the extension of the term of drug patents
- by Kim, Jin-Gu Mar 23, 2023 04:45am
- Previously, patentees could legally extend the duration of a patent unlimitedly, but the government's plan is to promote it up to 'up to 14 years'. According to the pharmaceutical industry on the 21st, the Korean Intellectual Property Office recently prepared an amendment to the Patent Act to reform the system for the duration of drug patent rights. The amendment is expected to be submitted to the National Assembly through legislative acts. In the current drug patent term extension system, there is no separate upper limit (cap). In addition to the essential patent period of 20 years, it extends the time taken for clinical trials and approval and approval of drugs. In other words, the duration of drug patents can be 21 years, 27 years, or 37 years in the '20+α' method. However, the United States and Europe set an upper limit on the additionally recognized period. This means that even if it took a total of 23 years for clinical trials, licensing, etc., only 14 years (USA) or 15 years (Europe) are recognized. On the other hand, Korea and Japan do not have a separate upper limit for this period. As a result, criticism has been constantly raised in the pharmaceutical industry that the patent holder's patent term is excessively long. As part of the evergreening strategy, the original company takes a strategy of registering a new patent and extending the total duration before the patent expiration date. It is criticism.
- Company
- Forxiga's generics will be suspended until the 7th
- by Kim, Jin-Gu Mar 22, 2023 05:47am
- Manufacturing and sales of Dong-A ST's SGLT-2 inhibitor-type diabetes treatment Dapapro are expected to be suspended by early next month. According to the pharmaceutical industry on the 20th, the Seoul Central District Court recently cited AstraZeneca's request for an injunction to prohibit infringement of patent rights filed against Dong-A ST. As a result, Dong-A ST will not be able to manufacture and sell Dapapro until the 7th of next month, when Forxiga's patent expires. Dapapro is a follow-on drug to Forxiga. Among the SGLT-2 inhibitors of Dapagliflozin, the original diabetes treatment, it is currently the only one listed as reimbursement, except Forxiga. In November of last year, Dong-A ST succeeded in avoiding part of the duration of the material patent for Forsyga alone in the first trial by using a 'prodrug' strategy. With this decision, Dong-A ST obtained the right to release a follow-on drug before the patent expiration of Forxiga. In December, Forxiga released Dapapro, a follow-up drug, for reimbursement. It was five months before the expiration of the Forxiga patent. AstraZeneca appealed to the Patent Court immediately after the first trial. At the same time, it filed an application for a provisional injunction requesting a ban on infringement of Dong-A ST's patent rights, including the manufacture and sale of Dapapro, until the second trial verdict. The Seoul Central District Court cited AstraZeneca's application for a provisional injunction. An official from AstraZeneca Korea said, "We welcome the decision of the Seoul Central District Court. Substance patents for active ingredients must be respected for the development of the pharmaceutical industry in Korea." We will work hard to do so,” he said.
- Company
- 10 years of the global launch of K-biosimilars
- by Chon, Seung-Hyun Mar 22, 2023 05:47am
- The cumulative exports of biosimilar products developed by Celltrion and Samsung Bioepis recorded 13 trillion won. Celltrion's Remsima is rapidly expanding its territory 10 years after entering the European market. Celltrion's biosimilars posted cumulative exports of 9 trillion won, and Samsung Bioepis' overseas sales of more than 4 trillion won. ◆Celltrion Health, accumulated exports of 9 trillion won by selling 4 similar types since 2013 According to the Financial Supervisory Service on the 22nd, Celltrion Healthcare's four biosimilars, Remsima, Remsima SC, Truxima, and Herzuma, posted a total of 1.71 trillion won in exports. It increased by 9.2% from the previous year and broke the record for the largest export. Celltrion Healthcare is an affiliate of Celltrion, and Celltrion Healthcare Holdings is the largest shareholder (24.3% stake). Celltrion Healthcare receives antibody biosimilar products from Celltrion and sells them to global distributors. Celltree Healthcare is selling four biosimilars in overseas markets: Remsima, Remsima SC, Truxima, and Herzuma. Remsima is a biosimilar product of Remicade. Remsima SC is a subcutaneous formulation of Remsima. Truxima and Herzuma are biosimilar products of anticancer drugs Mabthera and Herceptin, respectively. Last year, Remsima recorded the largest export of 859.3 billion won. It increased by 6.1% from 809.6 billion won in 2021 and produced the most exports since obtaining European permission in 2013. Despite intensifying competition in biosimilars, Remsima continues to increase exports as more than 100 countries obtained item approval last year. Remsima recorded exports of 145.3 billion won in 2013, and the cumulative amount of exports for 10 years until last year totaled 5,163.1 billion won. Remsima SC exported 236.9 billion won last year, more than double the previous year's 89.6 billion won. With only two products, Remsima and Remsima SC, the joint venture achieved 1.96 trillion won in exports last year. Remsima SC made its first export of 34.8 billion won in 2020 and recorded a cumulative export performance of 361.4 billion won over the past three years. Truxima showed exports of 436.5 billion won last year, down 4.9% from the previous year. It is the second consecutive year of decline since recording 786.8 billion won in 2020. It is analyzed that the growth rate has slowed somewhat as the competition for biosimilars intensified. Truxima posted its first export of 383.2 billion won in 2017 and achieved a total of 2.6148 trillion won in overseas sales over the six years until last year. Herzuma's export volume last year was 181.7 billion won, down 13.9% from the previous year. After posting overseas sales of 211 billion won in 2021, the growth trend has slowed down. Herzuma recorded a cumulative export performance of 865.6 billion won from 2017 to last year. Celltrion's four biosimilars jointly exported a total of 9.49 trillion won for 10 years from 2013 to last year. Samsung Bioepis is also breaking new records every year. Samsung Bioepis' operating profit was 231.5 billion won, up 20.1% from the previous year, and sales were 946.3 billion won, up 11.7% from the previous year. Both operating profit and sales are the highest since its launch in 2012. Sales increased by 21.7% in two years from 777.4 billion won in 2020, and operating profit increased by 59.6% during the same period. The operating profit to sales ratio last year was 24.5%, the highest ever. Starting with the Enbrel biosimilar in January 2016, Samsung Bioepis received approval for six and five biosimilars in Europe and the US, respectively. Biosimilars of five biosimilars of Samsung Bioepis, Enbrel, Remicade, Humira, Herceptin, and Lucentis, have been approved in Europe and the United States. In Europe, it has additionally obtained approval for the sale of Avastin biosimilars. Samsung Bioepis is selling five biosimilars in Europe and three biosimilars in the US. Samsung Bioepis started selling Enbrel and Remicade biosimilars in Europe in 2016. In 2018, it introduced biosimilars to Herceptin and Humira markets and started selling Avastin biosimilar AYBINTIO in Europe in 2021. In the US, among the five licensed products, Remicade biosimilar RENFLEXIS was launched in the US market in 2017, and Herceptin biosimilar Ontruzant was launched in the US in 2020. Last year, it started selling Lucentis biosimilar BYOOVIZ in the US. Established in 2012, Samsung Bioepis generated sales of 43.7 billion won for the first time in 2013. In 2016, when the biosimilar overseas targeting began in earnest, it recorded sales of 147.5 billion won, and has continued to grow every year since. Since its launch in 2012, Samsung Bioepis has recorded cumulative sales of 4,311.2 billion won. Most of Samsung Bioepis' sales come from overseas sales of biosimilars or royalties. Domestic sales volume is negligible. According to IQVIA, a pharmaceutical research institute, Samsung Bioepis' five biosimilars sold a total of 42.5 billion won last year. In other words, the biosimilars of Celltrion and Samsung Bioepis have exported more than 13 trillion won in total over the past 10 years.
- Company
- Dupixent likely to expand reimb to pediatrics from April
- by Eo, Yun-Ho Mar 22, 2023 05:46am
- Reimbursement of the atopic dermatitis treatment ‘Dupixent’ is expected to be expanded for pediatric and adolescent patients from April. According to sources from related industries, Sanofi-Aventis Korea recently concluded a drug price negotiation with the National Health Insurance Service for the low dose (200mg) of Dupixent (dupilumab). Thus, Dupixent will be presented to the Health Insurance Policy Deliberation Committee tomorrow (23rd). On the 23rd, JAK inhibitors such as Abbvie Korea’s ‘Rinvoq (upadacitinib)’ and Pfizer Korea’s ‘Cibinqo (abrocitinib)’ will also be discussed. Unless otherwise, treatment options for pediatric atopic dermatitis patients will be drastically expanded in April. Dupixent's journey to expand reimbursement has seem many twists and turns. Being an RSA (Risk Sharing Agreement) drug, and having added a separate dose of 200mg, Dupixent had to go through an additional cost-effectiveness review procedure with the Health Insurance Review and Assessment Service, and complete drug price negotiation with the National Health Insurance Service. If approved, Dupixent will finally see results after two years since applying for an expansion of reimbursement in April 2021. Although the specific indications may differ, the difference in speed of progress is evident when compared to JAK inhibitors such as Rinvoq and Cibinqo, which also have applied for expansion of reimbursement in atopic dermatitis. The price of JAK inhibitors are relatively lower than that of Dupixent. Rinvoq and Cibinqo were both listed for reimbursement in May last year and Rinvoq is also attempting to expand its reimbursement to pediatric adolescent patients. Meanwhile, Dupixent is the first and only biologic to specifically target and inhibit the signaling of IL-4 and IL-13, which is the fundamental cause of atopic dermatitis. As Dupixent does not have a black box warning from the US FDA, which addresses the most severe side effects, and does not require monitoring for organ toxicity during administration, it was approved without any requirement for testing when starting treatment or regular safety monitoring during treatment.
- Company
- Patent extension system for drugs to be reorganized…
- by Kim, Jin-Gu Mar 22, 2023 05:46am
- A plan to reorganize the patent term extension system of biopharmaceuticals in a manner similar to those of the US and Europe is being reviewed. In the pharmaceutical industry, it is predicted that if the reorganization plan passes the National Assembly, the patent term of some original drugs will be reduced to about one year. According to the pharmaceutical industry, the Korean Intellectual Property Office recently prepared an amendment to the Patent Act to reform the system for the patent term of drugs on the 21st. The amendment is expected to be submitted to the National Assembly in the form of legislative acts. The main point of the amendment is to set the upper limit on the remaining (valid) patent term to 14 or 15 years from the time a new drug receives approval. The pharmaceutical industry's attention is focused on how much the original drug's patent term will be shortened. In this regard, the US and Europe have systems that limit the effective patent term. This method allows the patent to be effective for up to 14 or 15 years from the time the drug has been approved. For example, even if a pharmaceutical company 'A' has been granted approval for a patent for 20+5 years (basic patent term + extended period), the entire patent term is shortened 'up to 15 years from the time of approval' by setting an upper limit. On the other hand, Korea does not have a separate limit on the patent term that is applied from the time of approval. In the current system, there are only regulations that limit the extension of the patent term to a maximum of 5 years, but no upper limit on the effective patent term. As a result, the Korean patent term of pharmaceuticals is longer than the US and Europe, with the same product and same patent. Taking Pfizer’s ALK-targeted anti-cancer drug Xalkori (ingredient: crizotinib) as an example, its remaining patent term is limited to 14 years after approval by the FDA in the US. Under the upper limit, only 1 year and 6 months (547 days) of the patent term extension was recognized for Xalkori in the United States. The situation is similar in Europe. Europe has an upper limit of 15 years from the date of initial marketing authorisation. As a result, only 2 year and 2 months (799 days) of the patent term extension was recognized for Xalkori in Europe. Since there is no separte regulation that states the upper limit in Korea, the extended patent term of Pfizer's Xalkori was fully recognized. The extended patent term approved for Xalkori in Korea is 2 years and 10 months (1034 days), which is about 8 months longer that that of Europe and about 16 months longer than that of US. If the amendment passes the National Assembly, the effective patent term of the original drug will be shortened to about 1 year. In other words, the timing of generic release in Korea will be accelerated. The key is whether the amendment will be applied only to newly introduced drugs or to existing drugs as well. If the amendment is applied to existing drugs, quite a number of drugs will be affected by the new system. According to the Korean Intellectual Property Office, as of the end of 2021, 360 drugs still have effective patent term remaining in Korea. In terms of number of patents, there are 612. Among them, 60 drugs (83 patent rights) are expected to become subject to the new system, having 'effective patent term of 15 years or more.' While the Korean Intellectual Property Office is reportedly considering the 14-year regulation, the US model, the pharmaceutical industry predicts that the effective patent term for 40 to 50 drugs may be shortened if the amendment is applied to existing drugs as well.
- Company
- Pediatric indication becomes key to sales in Korea
- by Moon, sung-ho Mar 21, 2023 05:56am
- The treatment market for pediatric and adolescent patients have grown rapidly last year. In particular, injections for pediatric and adolescent patients have gained much popularity and raised sales in reimbursed and non-reimbursed markets in hospitals and clinics. The transition from the COVID-19 pandemic to an endemic had increased focus on injections for obesity and growth, which led to great popularity. If the pediatric ‘non-reimbursed’ market had achieved great growth last year, this year, the market for reimbursed treatment for 'severe' pediatric conditions is expected to grow rapidly because major treatments for pediatric patients are attempting reimbursement in the first half of this year. First, Saxenda (liraglutide), the leader in the domestic obesity treatment market, saw rapid growth in sales last year with its expansion to the pediatric indication. #According to the market research institution IQIVA, Novo Nordisk’s Saxenda raised sales of KRW 58.9 billion last year, which is a 62.7% YoY increase. Saxenda’s sales had previously fallen for 2 consecutive years. From KRW 42.6 billion in 2019, sales had fallen to KRW 36.8 billion in 2020, then to KRW 36.2 billion in 2021. However, with COVID-19’s transition to an endemic, its sales had again surged and dominated Korea’s obesity treatment market. Saxenda’s sole lead in the market is expected to continue until new obesity treatments like Wegovy (semaglutide, Novo Nordisk), and Mounjaro (tirzepatide, Lilly) are released to the market. Saxenda’s rapid growth in sales is in line with its indication extension to children and adolescents last year. In December 2021, Saxenda’s indication was extended to treat adolescent patients aged 12 to 18 years with a BMI that corresponds to 30 kg/m2 for adults and weighs over 60 kg in Korea. In other words, the indication expansion to pediatric adolescent patients maximized sales in obesity and growth clinics and hospitals in Korea. The same goes for the growth hormone market. The growth hormone injection market, which had previously consisted of products from domestic pharmaceutical companies, has also grown into a large non-reimbursed treatment market and was evaluated to be sized at KRW 250 billion won last year with rising sales from growth clinics at front-line hospitals and clinics. If the pediatric ‘non-reimbursed’ market had achieved great growth last year, reimbursement extensions of major treatments for major pediatric conditions are expected this year. Key products include Dupixent (dupilumab) and Hemlibra (emicizumab). Both drugs received recognition for their need to extend reimbursement by the Health Insurance Review and Assessment Service’s Drug Reimbursement Evaluation Committee and are undergoing drug pricing negotiations with the National Health Insurance Service. If the pricing negotiations between the NHIS and pharmaceutical companies progress smoothly, the drugs will be able to receive reimbursement extensions within the first half of the year. In the case of Dupixent, if reimbursement extensions are finally made to cover children and adolescents with atopic dermatitis in May, its sales are expected to continue to grow as in the previous year. In the case of Dupixent, sales increased 35% YoY from the KRW 77.2 billion in the previous year to reach KRW 104, but experts predict there is still room for additional growth. The same goes for JW Pharmaceutical’s Hemlibra. Hemlibra is currently only reimbursed to treat patients who show resistance and have antibodies to existing treatments. As most of the Type A hemophilia patients are ‘non-antibody’ patients, this means that only a very few patients are receiving reimbursement for Hemlibra. This is why parents of hemophilia A patients had been continuously raising demand for Hemlibra's reimbursement extension. According to the 2019 Hemophilia White Paper, over 90% of the 1,746 patients with hemophilia A in Korea, 1,589 patients, are non-antibody patients. Drug pricing negotiations are underway after HIRA made the decision to extend reimbursement, and if the negotiations are completed within the first half of the year, non-antibody patients will be able to use Hemlibra with reimbursement as well. Meanwhile, the financial authorities expect the health insurance claims amount to increase rapidly if Hemlibra’s reimbursement is extended to cover non-antibody patients. According to IQVIA, Hemlibra’s sales last year increased 5% YoY from the KRW 7.2 billion to reach KRW 7.6 billion last year. In other words, if reimbursement is extended to cover non-antibody patients, the drug may grow to become a large blockbuster product that brings in millions of won every year. Moreover, the pharmaceutical industry predicts that the drug will become a 'dark horse' in Korea’s hemophilia treatment market which is currently led by GC Pharma if Hemlibra’s reimbursement is extended to cover non-antibody patients. A health insurance official said, “In the case of hemophilia, around 10% are antibody patients, and 90% non-antibody patients. Therefore, if reimbursement for Hemlibra is extended to cover non-antibody patients, the drug’s sales will naturally increase significantly.”
- Company
- Increasing male infertility
- by Eo, Yun-Ho Mar 21, 2023 05:56am
- As male infertility increases, interest in follicle-stimulating hormone preparations is increasing. So far, women's infertility treatment has been more active than male infertility treatment. 93.4%of the cost of infertility, which was carried out by 2021, was used for female infertility patients, and only 6.6%were used for male infertility patients. Infertility can occur in both men and women due to deficiency of sperm formation due to male sperm, mobility, and low pre -stimulating hormonal glands, or ovarian function of women, closure of fallopian tubes, and endometriosis. According to the Social Society of Society of Propaganda (SRS), the cause of infertility is about 40% of male factors. Looking at the number of infertility patients in domestic men, it can be seen that about 43%increased from 6,2468 to 89,350 in five years from 2017 to 2021. On the other hand, female infertility patients increased by about 11% over five years. The reason for the recent increase in male patients who are diagnosed with infertility is that the perception that there may be a cause of infertility is expanded compared to the previous one. Kim Soo -woong, a professor of urology medicine at Seoul National University Hospital, said, “Infight is that infertility is a problem of all couples, and interest in the cause and treatment of male infertility seems to be increasing. "I need a treatment option and research on continuous male infertility treatment is needed." Infertility treatments, which are used in general, have a genital-stimulating hormone. The genital stimulating hormone components include Follicle Stimulating Hormone, Human Menopausal Gonadotropin (HMG), and Human Chorionic Gonadotropin preparations. Among them, the FHS formulation is a hormone that is synthesized and secreted in the pituitary gland, which is not only involved in producing eggs in the ovaries of women but also acts as a hormone that produces sperm in the testicles of men, thereby improving the reproductive function of infertility patients. Among the FSH preparations used in Korea, 'Puregon' is the only man who has an indication of the deficiency of sperm formation caused by men's low -stimulating hormonal glands. PureGon confirmed the effectiveness of the combination of HCG, which was conducted with HCG, which was a lowly stimulating hormonal sexual dysfunction that failed to treat infertility with HCG monotherapy. According to the guidelines of the Endocrinology Society, FSH+HCG combination therapy is essential for men who are reduced in the genitals of the central central central central central central central central central central central central cavity in the treatment of reproductive ability of men. Professor Kim said, “The infertility treatment environment is far from effective infertility through male infertility diagnosis and treatment, and it is increasing the burden on infertility treatment for women. "Everybody needs to be infertile and treated in an appropriate time regardless of gender." World Health Organization (WHO) defines as infertility a couple with a normal marital relationship without contraception who are not pregnant for a year. As of 2021, the number of male and female patients diagnosed with infertility in Korea was about 250,000, up 9% from 2019, and nearly 10,000 people per year for nearly five years. Recently, the city of Seoul also announced a plan to expand infertility support to solve the problem of low outflows.
- Company
- Pharmaceutical exports continue to be sluggish...
- by Kim, Jin-Gu Mar 20, 2023 05:51am
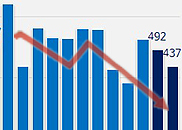
- Domestic drugs are continuing to show sluggish sales overseas. During the prolonged COVID-19 crisis, Korea enjoyed an uprise in pharmaceutical exports, but due to the nearing endemic, exports are returning to the pre-COVID-19 level. In particular, the sharp drop in exports of COVID-19 vaccines manufactured in Korea affected the decline in Korea’s overall export performance. According to the Korea Customs Service on the 17th, Korea's pharmaceutical export amounted to USD 928.49 million (approximately KRW 1.22 trillion, based on the won-dollar exchange rate of 1313.00 won). This is the lowest amount recorded in January and February alone over the past three years. It is also 16.5% lower than the USD 1.12 billion made in January and February of last year and 34.0% lower than the USD 1.47 billion made in January and February 2021. Domestic pharmaceutical exports had increased significantly during the prolonged COVID-19 period. Monthly exports prior to COVID-19 were around USD 300 million, however, after COVID-19 started to spread and become a pandemic in March, exports nearly doubled to average around USD 600 million per month. In December 2021, monthly exports soared to exceed USD 1 billion. The increase in confirmed COVID-19 patients increased the demand for medical care, and this is analyzed to have led to the increase in the export of domestic drugs. Also, from the end of 2021, exports of Moderna and Novavax vaccines that were manufactured in Korea began in earnest and contributed to the increase in exports. However, the slowdown in the spread of COVID-19 in the second half of last year then led to a decrease in the monthly exports of domestic drugs. In particular, this decrease in domestic pharmaceutical exports started in earnest in Q4 last year. Monthly drug exports, which consistently remained over USD 500 million until September last year, fell further to USD 428.51 million in October, then to USD 393.33 million in November. Although exports briefly increased to USD 525.97 million in December last year, the amount fell back to USD 400 million range entering this year. The industry believes that the decline in COVID-19 vaccine exports affected the decline in total exports. Overseas exports of domestic vaccines had soared from December 2021 to the first half of last year. Vaccine exports, which ranged between USD 10 million and USD 20 million a month before then, increased to exceed USD 270 million in December 2021, and then steadily maintained exports of over USD 100 million every month. However, after falling to USD 60 million in July last year, the amount has since returned to the standard level.
- Company
- Leclaza costs 88 billion won for phase 3 in 3 years
- by Chon, Seung-Hyun Mar 20, 2023 05:51am
- Last year, Yuhan invested a total of 88 billion won in phase 3 clinical trial of Leclaza, a new anti-cancer drug. Yuhan Corporation is challenging the first-line treatment approval based on the efficacy and safety of Leclaza confirmed in clinical trials. According to the Financial Supervisory Service on the 20th, as of the end of last year, Yuhan Corporation's development costs reflected as intangible assets totaled 104.8 billion won. It increased by 33.1 billion won in one year from 71.7 billion won at the end of 2021. In 2019, the Financial Supervisory Service set a standard that accounting assets can be treated only when there is technical feasibility of R&D tasks such as new drugs. The Financial Supervisory Service suggested that R&D costs can be turned into assets by initiating phase 3 clinical trials for new drugs and approval of phase 1 clinical trials for biosimilars. Generics can be capitalized after the BA test is approved. Yuhan's R&D intangible assets are R&D costs invested in Leclaza and eight IMDs. Among them, Leclaza's development cost intangible assets amounted to 88 billion won. A total of KRW 88 billion was used for Leclaza's phase 3 clinical trial. Leclaza accounted for 84.0% of Yuhan's development cost intangible assets. Leclaza is a non-small cell lung cancer treatment approved as the 31st new drug developed in Korea in January 2021. Patients with locally advanced or metastatic non-small cell lung cancer who developed T790M resistance after administration of first- and second-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) are eligible for the treatment. It acts as a mechanism to inhibit the proliferation and growth of lung cancer cells by interfering with the signal transduction involved in the growth of lung cancer cells. Leclaza recognized development costs of 32.6 billion won as intangible assets for the first time in the fourth quarter of 2020. As phase 3 clinical trials began in earnest, development costs were reflected as intangible assets. Leclaza's development cost intangible assets increased to 61.4 billion won at the end of 2021, and 26.6 billion won was added last year. As the cost of phase 3 clinical trials for Lecraza increased, the development cost that Yuhan Corp. reflected as an asset exceeded 100 billion won for the first time. Yuhan is challenging the first-line treatment approval based on the results of Leclaza's phase 3 clinical trial. On the 17th, Yuhan Corporation applied for approval to use Lexraza as a first-line treatment for patients with EGFR exon 19 deletion or exon 21 (L858R) substitution mutations in locally advanced or metastatic non-small cell lung cancer. Leclaza demonstrated superior safety and efficacy compared to existing treatments in phase 3 clinical trial (LASER301) conducted on 393 patients with active EGFR mutation-positive locally advanced or metastatic non-small cell lung cancer who had not previously received treatment. The clinical results were recently unveiled at the Asian Congress of the European Society for Oncology held in Singapore. If Leclaza's use as a first-line treatment expands, sales are projected to soar. Although Leclaza is used on a limited basis as a second-line treatment, it is evaluated that it has successfully settled in the market. According to IQVIA, a pharmaceutical research institute, Leclaza recorded sales of 16.1 billion won last year. It increased by 4 times from 4.1 billion last year. Leclaza entered the prescription market in earnest in July 2021 with its listing on the health insurance benefit list. Sales of KRW 1.5 billion and KRW 2.6 billion occurred in the third and fourth quarters of 2021, respectively. Leclaza sold 3.2 billion won and 3.7 billion won in the first and second quarters of last year, respectively, and expanded to 4.6 billion won and 4.5 billion won in the third and fourth quarters. Leclaza recorded cumulative sales of 20.2 billion won for a year and a half after its release. Leclaza has already broken the sales record for new anti-cancer drugs developed in Korea. Domestically developed anti-cancer drugs approved prior to Leclaza include Ilyang Pharm's Supect, Dongwha Pharm's Milican, Chong Kun Dang's Camtobell, Samsung Pharm's Riavax, and Hanmi Pharm's Olita. None of these products exceeded 10 billion won in annual sales.

- Company
- Possibility of passing the drug price reduction Act
- by Kim, Jin-Gu Mar 20, 2023 05:51am
- A sense of crisis in the biopharmaceutical industry is rising as the possibility of passing the 'drug price reduction/refund bill' in the plenary session of the National Assembly is increasing. The pharmaceutical industry is concerned that the amendment will infringe on the right to request a trial as stipulated in the Constitution, and furthermore, it will damage the domestic pharmaceutical and bio-industry as a whole. According to the pharmaceutical industry on the 16th, the revision of the National Health Insurance Act, the so-called Drug Price Reduction and Refund Act, proposed by Rep. Kim Won-i and Nam In-soon, respectively, will be decided at the National Assembly plenary session this month at the earliest. According to this amendment, if a pharmaceutical company loses a lawsuit on drug price cuts, it must discharge the health insurance finances invested during the drug price cut suspension period. In the opposite case, the amended bill also contains content that allows the NHIS to refund the damages incurred by the pharmaceutical company during the course of the lawsuit if the pharmaceutical company wins the primary case. However, in the pharmaceutical industry, the position is that in the case of 'refund', it does not fall under the requirements for citation for suspension of execution. In the current legal system, suspension of execution is cited when there is a 'concern about serious and irreparable damage'. However, in the case of a refund, it is argued that these requirements are not met. As a result, concerns are raised that the enforcement of the law will be biased toward the recovery among refunds and refunds, which will violate the right to request a trial specified in the Constitution and worsen the remedy of rights. There are concerns that companies' attempts to suspend the execution of the government's drug price cuts will significantly shrink. In the pharmaceutical industry, it is the position that the economic gains and losses caused by the difference between the decision to suspend execution and the judgment on the merits should be remedied according to the existing procedures, such as claims for damages between companies or parties. It is also criticized that it is not right in the legal system for the state to consider unfair gains and return them after the fact, and that excessive infringement of property rights is unconstitutional. Furthermore, the pharmaceutical industry is concerned that if this amendment is passed, it will deal a blow to the domestic pharmaceutical bio-industry as a whole. In fact, in past cases of suspension of execution citations, three-fourths of the total number of citations are for domestic pharmaceutical companies. The expected surcharge upon redemption of these is estimated at 490 billion won. This is eight times higher than multinational pharmaceutical companies' 59 billion won. In particular, the pharmaceutical industry is criticizing that legal disputes over drug price reduction, which is a legitimate right of companies, will shrink in a situation where government re-evaluation mechanisms such as re-evaluation of already-listed drugs, re-evaluation of benefit adequacy, and re-evaluation of overseas drug prices are expected to continue in the future. It is expected that the Drug Price Reduction Act will discourage domestic pharmaceutical companies from challenging patents and thereby worsen the positive function of reducing health insurance finances. This is because if the original company finally wins the lawsuit and the NHIS refunds the pharmaceutical company the loss caused by the drug price cut, the NHIS can claim the right of indemnity from the generic company that participated in the patent lawsuit. The pharmaceutical industry was concerned that the amendment would undermine generic companies' willingness to challenge patents and hinder active patent challenges. As a result, there are concerns that the opportunity for financial savings due to the release of generics will be lost.