- LOGIN
- MemberShip
- 2026-05-07 16:18:32
- Company
- Hanmi to examine the possibility of the Poseltinib effect
- by Chon, Seung-Hyun Jun 14, 2023 05:38am
- Hanmi Pharm is examining the possibility of new indications with new drug candidates returned by multinational pharmaceutical company Eli Lilly. Hanmi Pharmaceutical announced on the 12th that it announced the interim results of phase 2 clinical trial of three-drug combination therapy, a follow-up study of the BTK inhibitor Poseltinib, at the European Society of Hematology recently held in Frankfurt, Germany. The safety and efficacy of the three-drug combination therapy including Poseltinib in relapsed and refractory diffuse large B-cell lymphoma were confirmed. Hanmi Pharmaceutical and Genome Opinion supported it, and Professor Byun Ja-min of the Department of Hematology and Oncology at Seoul National University Hospital gave a presentation. Poseltinib is a BTK inhibitor that Hanmi Pharm transferred technology to Lilly in 2015 for up to $690 million, including a $50 million down payment. BTK inhibitors are drugs with a mechanism of inhibiting Bruton's Tyrosine Kinase protein, which plays a key role in B cell growth. Lilly returned the rights in January 2019, citing failure to prove efficacy in phase 2 clinical trials for patients with rheumatoid arthritis. In October 2021, Hanmi Pharmaceutical signed a contract with Genome Opinion to jointly develop Poseltinib. Afterward, Genome Opinion conducted a researcher-led clinical trial (GPL study) for relapsed and refractory DLBCL patients through a three-drug combination therapy combining Poseltinib, CD3xCD20 bispecific antibody Glofitamab, and immunomodulator Lenalidomide. In the interim results presented at this EHA, the research team confirmed the safety and effectiveness of GPL therapy. Of the 14 patients whose response was evaluated after the start of the clinical trial, 79% met the OR, which is the efficacy evaluation criterion. Despite the initial data, 36% of patients had CR with the disappearance of cancer cells. The safety cohort also had no specific adverse reactions. The research team evaluated that the GPL combination therapy has sufficient potential as a new treatment option for patients as it can broadly control the carcinogenesis of DLBCL compared to existing therapies. "We expect that it will be a treatment that gives new hope to relapsed and refractory DLBCL patients who have failed standard treatment, including CAR-T," said Professor Yoon Seong-soo of the Department of Hematology and Oncology at Seoul National University Hospital, coordinator of the entire clinical trial. said, “As the safety and effectiveness of Poseltinib and bispecific antibody combination therapy have been confirmed, we expect that the final clinical results will provide new treatment options to medical staff in the future.”
- Company
- Hanmi's NASH drug candidate, designated as a fast track
- by Jun 14, 2023 05:38am
- A researcher at Hanmi Pharm is conducting research on new drug candidates. (Photo: Hanmi Pharm)Hanmi Pharmaceutical announced on the 13th that Efinopegdutide, a new drug candidate for non-alcoholic steatohepatitis (NASH), has been designated as fast-track by the US Food and Drug Administration (FDA). Efinopegdutide is a dual agonist that simultaneously activates the GLP-1 receptor, which helps the secretion of insulin and suppresses appetite, and the glucagon receptor, which increases energy metabolism. The technology was transferred to MSD in August 2020. This fast-track designation was led by MSD. Fast Track is a system to expedite the screening of new drug candidates being developed targeting diseases with high unmet medical demand due to lack of treatment. Phase 2a clinical trial of Efinopegdutide, which was designated as a fast-track this time, was recently completed. MSD plans to orally present the results of phase 2a clinical trial of Efinopegdutide in adult patients with NAFLD at EASL, which will be held in Vienna, Austria from the 21st (local time) to the 24th. In addition to the phase 2a clinical trial, MSD plans to initiate an additional phase 2b clinical trial for Efinopegdutide within this month.

- Company
- Will a new treatment option be introduced for gastric cancer
- by Jung, Sae-Im Jun 14, 2023 05:38am
- Unlike lung cancer and breast cancer, gastric cancer has been regarded as one type of cancer that has not benefited from the development of new anticancer drugs. For the past decade, chemotherapy has been the standard first-line treatment for patients with HER2 gene-negative metastatic gastric cancer. This means that there were no suitable new drugs other than HER2-targeting anticancer drugs available for its treatment. However, changes have recently occurred in the treatment of gastric cancer. The immuno-oncology drug ‘Opdivo’ emerged as a first-line option, another immuno-oncology drug 'Keytruda' also obtained positive results. A targeted therapy that targets a new protein is also expected to enter the market. Two noteworthy studies in the field of gastric cancer were presented at the ‘2023 ASCO Annual Meeting (ASCO 2023)’ that was held for 5 days from the 2nd (local time). The two were: results from the Phase III trial of ‘zolbetuximab,’ a targeted anticancer drug that targets CLDN18.2 (Claudin 18.2), and results from a Phase III trial that reviewed the first-line treatment of Keytruda in gastric cancer. Primary endpoint results of Phase III GLOW trial on zolbetuximab (Data: ASCO) Zolbetuximab is a new drug being developed by Astellas that targets CLDN18.2 for the first time among anticancer drugs. Claudin 18.2 is a protein mainly present on the surface of gastric cancer cells. Although the protein is also present in normal cells, it is expressed at high levels in certain malignant tumors. Claudin 18.2 is known to be involved in the proliferation, differentiation, and metastasis of cancer cells. The GLOW clinical trial that was announced this year, compared zolbetuximab + CAPOX (capecitabine + oxaliplatin) combination therapy with just CAPOX in 507 patients with advanced·metastatic gastric cancer who were CLDN18.2 positive. Of those involved, 78% had a PD-L1 combined positive score (CPS) less than 5. Min-Hee Ryu, Professor of Oncology at Asan Medical Center The primary endpoint, median progression-free survival was significantly higher - 8.2 months - in the zolbetuximab group compared with the 6.8 months in the placebo group (HR=0.69). At 24 months, the PFS rate in the zolbetuximab group was 14%, twice higher than the 7% in the placebo group. The secondary endpoint, overall survival (OS) was 14.4 months in the zolbetuximab group and 12.2 months in the placebo group, demonstrating a 23% reduction in risk of death with zolbetuximab (HR=0.77). In addition, the objective response rate was 53.8% and the duration of response was 6.3 months in the zolbetuximab group. During an interview with Dailypharm, Min-Hee Ryu, Professor of Oncology at Asan Medical Center, said, “Zolbetuximab is expected to bring a positive effect as it has demonstrated superiority in OS as well as PFS. In terms of side effects, although the frequency of nausea and vomiting, which was different from those of existing drugs, was high with zolbetuximab, but appears to be at a manageable level. I believe the side effects arise because the targeted claudin protein is also present in normal cells.” Another noteworthy clinical trial is the Phase III KEYNOTE-859 that was conducted to evaluate Keytruda’s effect as a first-line treatment for gastric cancer. This clinical trial is significant in that MSD succeeded in changing the design of the KEYNOTE-062 clinical trial, which had previously failed in the first-line, and was led by Korean medical staff. The clinical trial compared Keytruda with the chemotherapy CAPOX or 5-FU (5-fluorouracil) to chemotherapy alone in patients with HER2-negative gastric cancer. The trial also measured the therapy’s effect according to the patient’s PD-L1 CPS score. The primary endpoint was overall survival (OS). Primary endpoint results of KEYNOTE-859 study on Keytruda as first-line therapy in gastric cancer (Data: ASCO) Trial results showed that the median overall survival (OS) of the Keytruda group was 12.9 months, reducing the risk of death by 22% compared with placebo (HR=0.78). Keytruda’s effect increased with the increase in CPS. The hazard ratio was 0.83 in the patient group with a CPS score of 1 to 10, and the hazard ratio was 0.64 in the patient group with a CPS score of 10 or more. This means that in patients with a CPS score of 10 or higher, Keytruda lowered the risk of death by 35%. In addition, the Keytruda group demonstrated significant efficacy in secondary key endpoints as well, including PFS. Sun-Young Rha, Professor of Oncology at Yonsei Cancer Hospital, Another immuno-oncology drug, 'Opdivo,' had proven its efficacy in patients with a CPS score of 5 or higher and obtained an indication for the first-line treatment of gastric cancer. Although Keytruda succeeded in demonstrating its effect later than Opdivo, it is meaningful as the results included PD-L1 CPS 1-4 point patients. Sun-Young Rha, Professor of Oncology at Yonsei Cancer Hospital, said, “The fact that Keytruda showed an improvement with a hazard ratio of 0.83 even in patient groups that include patients with CPS 1 to 4, demonstrates that patients with a CPS of 1 or more may use the immunotherapy.” They agreed that it will become necessary to devise an effective treatment strategy according to the CPS score when zolbetuximab is introduced in the future. Professor Ra said, "Our task for the future in gastric cancer is to reach a consensus on how to treat patients who are both CLDN18.2 positive and PD-L1 positive." Professor Rhu added, “There is some overlap between the areas for the use of zolbetuximab and immuno-oncology drugs. in patients with CPS between 5 to 10, the effects of the two drugs are likely to be similar, so the side effect aspect should be considered. For those with a CPS of 10 or higher, immuno-oncology drugs are definitely more effective."
- Company
- Rozanolixizumab has been designated as an orphan drug
- by Eo, Yun-Ho Jun 14, 2023 05:37am
- Rozanolixizumab, a new drug for generalized myasthenia gravis, has been designated as an orphan drug. The Ministry of Food and Drug Safety recently announced that it designated UCB's gMG treatment Rozanolixizumab as an orphan drug. The target indication is the treatment of generalized myasthenia gravis in patients who are anti-acetylcholine receptor or anti-muscle-specific tyrosine kinase antibody positive and require additional therapy with corticosteroids or non-steroidal immunosuppressants. This drug was designated for expedited review by the U.S. FDA in January and is currently undergoing approval procedures in Japan. Rozanolixizumab is a type of monoclonal antibody for subcutaneous injection that targets the emerging Fc receptor (FcRn) and has proven its effectiveness through phase 3 clinical trials. Myasthenia gravis is a rare disease that affects about 5 per 100,000 people, and can occur in people of all ages. In children and the elderly, symptoms of decreased muscle strength are caused by diseases of the immune system. In 60% of patients with this disease, symptoms begin in the eye muscles. In particular, ptosis, in which the eyelids droop, and double vision, in which objects are seen twice, may occur. Systemic myasthenia gravis is an unpredictable chronic autoimmune disease, and as autoantibodies that induce the onset can target specific proteins in the postsynaptic membrane and impair synaptic transmission in the neuromuscular junction, major countries are developing candidates. It is designated as an expedited review target and orphan drug. Looking at the current status of myasthenia treatment development, Argenx's Efgartigimod alfa has obtained US FDA approval. Nipocalimab from Johnson & Johnson is also close to commercialization. In addition, Batoclimab, which received technology transfer from Hanall Biopharma, a domestic company for Immunovant, also completed phase 3.
- Company
- Ildong makes initial lead in the SGLT2·DPP4 combo market
- by Kim, Jin-Gu Jun 14, 2023 05:35am
- Pic of Qtern, Zemidapa, Esgliteo, and Sugadapa (clockwise from the upper left corner) Full-fledged competition has begun in the ‘SGLT-2 inhibitor+DPP-4 inhibitor’ two-drug combination therapy market for the treatment of diabetes. In the first month after 5 products were launched, outpatient prescriptions reached KRW 300 million, and AstraZeneca and Ildong Pharmaceutical's Qtern led the market bringing in prescriptions of KRW 200 million. This market is expected to expand further after the term expires for Januvia (sitagliptin) in September. Fierce competition is expected after the 86 generic companies release their two-drug combination of dapagliflozin + sitagliptin at the same time after September. According to the market research institution UBIST, SGLT-2i+DPP-4i two-drug combination therapies have posted KRW 300 million in outpatient prescription sales in May this year. The government had extended insurance reimbursement to cover three-drug SGLT-2i+DPP-4i+metformin combinations in April. With the approval, pharmaceutical companies have been applying for the reimbursement of their SGLT-2i+DPP-4i two-drug combination therapies. As of May 1st, 5 products, ▲Chong Kun Dang’s Exiglu-S Tab (dapagliflozin+sitagliptin); ▲AstraZeneca’s ‘Qtern Tab (dapagliflozin+saxagliptin); ▲Boehringer Ingelheim’s Esgliteo Tab (empagliflozin+linagliptin); ▲MSD’s Stegluzan Tab (ertugliflozin+sitagliptin); ▲LG Chem’s Zemidapa (dapagliflozin+gemigliptin), were listed for reimbursement. Front-line hospitals and clinics have been prescribing these products by adding the single-agent metformin to the two-drug combinations. In the first month after reimbursement listing, Qtern recorded the most prescriptions with sales of KRW 192 million among the 5 newly listed products. Zemidapa followed with KRW 66 million, and then Exiglu-S with KRW 22 million. Monthly prescriptions of Esgliteo and Stegluzan were less than KRW 20 million. The variable in the market is the coming entry of its latecomers. Over 90 companies will engage in competition by the end of the year. Dong-A ST’s Sugadapa (dapagliflozin+evogliiptin) has already entered the competition this month. Sugadapa was listed for reimbursement from the 1st of this month. Dong-A ST started the sale of its product in May without reimbursement in advance to offset the weakness of entering the market a month later than its competitors. With the addition of Dong-A ST, the number of competitors in this market has expanded to 5 domestic companies and 3 multinational pharmaceutical companies. In addition to LG Chem, Chong Kun Dang, and Dong-A ST are exclusively marketing their products. On the other hand, Boehringer Ingelheim has signed an agreement with Yuhan Corp to co-promote and sell its drug, and MSD with Chong Kun Dang. AstraZeneca’s Qtern is exclusively sold in Korea by Ildong Pharmaceutical. Also, many generics are expected to join the market from September this year. Generic companies are expected to release generics of the dapagliflozin+sitagliptan combination at the same time after Januvia's patent expires in September. 86 pharmaceutical companies have currently received approval for their generic versions of dapagliflozin+sitagliptan. The patent term for dapagliflozin (Forxiga), another drug, had expired in April. Daewoong Pharmaceutical's combination drug that contains its Envlo (enavogliflozin) is also expected to be released next year. Daewoong Pharmaceutical is developing a combination drug with Zemiglo (gemigliptin) to combine with its Envlo. Daewoong Pharmaceutical plans to release its combination product by Q1 next year. Some are predicting that the SGLT-2 inhibitor + DPP-4 inhibitor two-drug combination will not produce as good results as expected. As the addition of metformin is required for the reimbursement of the two-drug combination, the criticism is that the advantage of convenience in intake offered by combination drugs has faltered somewhat. In fact, prescriptions with the new products have no significant difference from the previous prescriptions that added a single DPP-4 inhibitor to SGLT-2 inhibitor+metformin two-drug combinations. Therefore, industry analysis is that the SGLT-2 inhibitor+metformin combination, which was previously released in bulk with Forxiga's patent expiry, will become mainstream in the prescription market.
- Company
- Discussions on expanding benefit for Jakavi's GVHD are slow
- by Eo, Yun-Ho Jun 13, 2023 05:43am
- Discussions on expanding insurance coverage for Jakavi's Graft-versus-Host Disease (GvHD) indication have been sluggish. According to the industry, Novartis Korea Jakavi passed the harmaceutical Reimbursement Criteria Subcommittee of the Health Insurance Review and Assessment Service last February, but it has not been submitted to the Financial Impact Assessment Subcommittee, which is a step before the Pharmaceutical Reimbursement Evaluation Committee. In May 2022, after obtaining indications for graft-versus-host disease in Korea, the drug submitted an application for an extension of coverage. However, after being put on hold for more than 8 months, discussions on salary have begun. GvHD is a serious complication that can occur after an allogeneic hematopoietic stem cell transplant (allo-SCT). The transplanted donor's T cells recognize the patient's normal cells as foreign substances and attack them, affecting various organs such as the skin, gastrointestinal tract, liver, and lungs. As symptoms can appear throughout the body, GvHD poses another challenge to patients who have survived allogeneic hematopoietic stem cell transplantation and affects their quality of life. Steroids are used as the first-line therapy, but about 50% of them fail, and in these cases, standard treatment has not yet been established, so there is an unmet demand for effective treatment options. Jakavi is an option for the treatment of patients with acute or chronic graft-versus-host disease aged 12 years or older who do not respond adequately to prior corticosteroid therapy in these circumstances. Kim Hee-jae, chairman of the Korean Society for Hematopoietic Stem Cell Transplantation (Professor of Hematology at Seoul St. Mary’s Hospital), said, “Jakavi has shown good effects through clinical studies in the treatment of acute and chronic patients, and similar results have been shown in actual practice. “It opens up new possibilities for many patients,” he said. Jakavi has proven its efficacy in GvHD through the phase 3 REACH2 study. As a result, the overall response (OR) on day 28 of the Jakavi-administered group was 62% (96/154), which was higher than that of the control group, Best Available Therapy (BAT)-administered group, 39% (61/155). On the 56th day, the durable overall response was confirmed to be 40% (61/154), about twice as high as 22% (34/155) of the control group. More about this source texture text is required for additional translation information.
- Company
- Severe asthma Tx Tezpire to soon be commercialized in Korea
- by Eo, Yun-Ho Jun 13, 2023 05:43am
- The new drug for severe asthma, ‘Tezpire' is soon to enter the Korean market. According to industry sources, AstraZeneca Korea has submitted an application for the approval of ‘Tezspire (tezepelumab)’ in Q2 to the Ministry of Food and Drug Safety and is receiving review. At this pace, Tezspire is expected to be commercialized in the second half of the year. As a viable competitor to Sanofi’s ‘Dupixent (dupilumab),’ Tezspire inhibits the action of the thymic stromal lymphopoietin (TSLP), a key epithelial cytokine that sits at the top of multiple inflammatory cascades, to block the inflammatory chain reaction. The drug was approved by the US FDA in 2021 as a treatment for adult and pediatric patients aged 12 years and older with severe asthma., and added a self-injection formulation to its approval in February this year. Tezspire demonstrated a significant reduction in asthma exacerbation in the Phase II PATHWAY trial and Phase III NAVIGATOR trial, which included a broad population of severe asthma patients irrespective of key biomarkers, including blood eosinophil counts, allergic status, and fractional exhaled nitric oxide (FeNO). The most common adverse reactions shown in those that received Tezspire in the trials were pharyngitis, rash, arthralgia, and injection site reactions. The findings from the NAVIGATOR study were previously published in The New England Journal of Medicine. The Korea National Enterprise for Clinical Trials had selected Tezspire as the No.1 drug in need of an urgent introduction to Korea in its report on the ‘List of foreign new unintroduced drugs that should be promptly Introduced to Korea.’
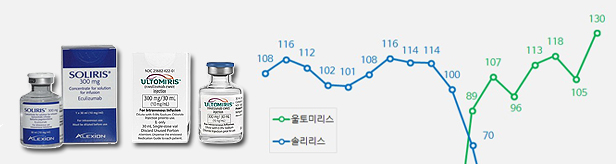
- Company
- Sales of Soliris and Ultomiris rise despite changes
- by Kim, Jin-Gu Jun 13, 2023 05:43am
- Soliris(left) and Ultromiris Quarterly sales of the rare disease treatments ‘Soliris (eculizumab)’ and ‘Ultomiris (ravulizumab)’ had risen significantly in Q1 this year. Although sales of Soliris remained similar to last year, sales of Ultomiris, its follow-on drug, soared in the same period. The analysis is that the generation change between the two drugs has neared completion. According to the market research institution IQVIA on the 12th, sales of Ultomiris in Q1 were KRW 13 billion, up 35% from KRW 9.6 billion in Q1 last year. During the same period, Soliris’s sales slightly fell from KRW 2.8 billion to KRW 2.4 billion. Soliris is a rare disease treatment indicated for the treatment of ▲ paroxysmal nocturnal hemoglobinuria, ▲ atypical hemolytic uremic syndrome (aHUS), ▲generalized myasthenia gravis, and ▲ neuromyelitis optica spectrum disease. Ultomiris is a follow-on of Soliris. As it has a half-life that is 4 times longer than that of Soliris, the drug may be administered every 8 weeks. However, the indication for the drug is limited to paroxysmal nocturnal hemoglobinuria/ Both drugs were introduced to Korea by Handok from Alexion. Soliris was approved in January 2010 and applied for reimbursement in 2018. Ultomiris was approved in May 2020 and its reimbursement was applied from June 2021. The combined sales of the two drugs peaked at KRW 15.9 billion in Q3 2021 and then was on a gradual decline. In Q4 last year, its sales decreased to KRW 12.9 billion. However, sales showed a rebound in Q1 this year, recording combined sales of KRW 15.4 billion. Quarterly Sales of Soliris·Ultromiris (Unit: KRW 100 mil, Data: IQVIA) The sales rebound received more attention because the domestic sales rights for the two drugs were transferred from Handok to AstraZeneca in Q1 last year. AstraZeneca acquired Alexion in 2020 for USD 39 billion (approximately KRW 42 trillion). After the sales rights agreement between Handok and Alexion expired in January this year, AstraZeneca Korea retrieved the sales rights to sell the two rare disease drugs directly in Korea. However, the prevailing opinion in the industry is that the sales right transfer had little or limited effect on the sales rebound. AstraZeneca established a rare disease Business Unit at the end of last year and completed the composition of the business unit in February of this year in time for the retrieval of Soliris and Ultomiris’s sales rights. The company was able to start full-scale marketing activities only in March. Therefore, the sales rebound could be rather interpreted as performance derived under the strong influence of Handok. Industry experts believe that the impact of the sales right transfer would only be measurable after Q2.
- Company
- Beova can be prescribed at general hospitals
- by Eo, Yun-Ho Jun 12, 2023 05:42am
- Beova, a new drug introduced by Jeil Pharmaceutical, will be available for Rx at general hospitals. According to related industries, Beova, an overactive bladder treatment, is currently available at 22 medical institutions nationwide, including SMC and AMC, as well as advanced general hospitals, Gangnam Severance Hospital, Sangnam Sacred Heart Hospital, Dongtan Sacred Heart Hospital, Chungnam National University Hospital, and Hanyang University Hospital (headquarters, Guri). Beova is a new drug introduced by Jeil Pharm from Japan's Kyorin Pharmaceuticals in November 2019. It obtained domestic development, manufacturing, and sales rights, and imported raw materials to produce finished products in domestic factories. In order to obtain domestic approval, Jeil Pharmaceutical conducted a phase 3 bridging clinical trial to compare and evaluate the safety and effectiveness of Beova in 210 overactive bladder patients at 20 domestic institutions, including Seoul Asan Medical Center, from May 2020 to this year. As a result, it was confirmed that the change in average urination frequency per day at 12 weeks from baseline as the primary efficacy evaluation variable was improved by -2.38 times compared to -1.22 times with a placebo. In the secondary endpoints, the average number of urinary urgency per day, the number of urge incontinence, the change in the number of urinary incontinence, and the change in the average voiding volume per time, there was a significant improvement compared to the placebo. Beova is a new β3-adrenergic receptor agonist that selectively acts on sympathetic nerve receptors to relax the bladder detrusor muscle, helping to treat symptoms such as frequent urination, urgency, and urgency urinary incontinence. Mirabegron was the only β3-adrenergic receptor agonist currently on the market, but the launch of Beova is expected to form a competitive landscape. Vibegron is known to be more than 9000 times more selective for β3 receptors than β1 and β2 receptors. In addition, Vibegron's maximum response rate for β3 receptors is 99.2%, which is higher than 80.4% for Mirabegron, the same β3 agonist. It is an ingredient that has the advantage of having a low risk of cardiovascular side effects because it does not stimulate receptors. It is known that there is very little concern about interactions with drugs that pass through the CYP2D6 metabolic pathway and that it can be administered in normal doses even to patients with hepatic or renal impairment. Lee Gyu-seong, professor of urology at SMC, said, "Beova is a new drug that is introduced in Korea. It will help provide high-quality treatment effects to overactive bladder patients in Korea with excellent symptom improvement and low adverse reaction rates."
- Company
- Eisai applies for approval of its AD drug lecanemab in KOR
- by Jung, Sae-Im Jun 12, 2023 05:42am
- Pic of lecanemab The drug that is being considered the next blockbuster candidate, Eisai and Biogen’s Alzheimer’s treatment ‘lecanemab,’ is set to enter the Korean market. Eisai announced on the 8th that it had submitted an application for the marketing authorization of lecanemab to the Ministry of Food and Drug Safety to treat mild cognitive impairment or mild dementia stage of disease arising from Alzheimer’s disease (AD). This is the third application the company had filed in Asia after Japan and China. Lecanemab was jointly developed by the two companies. Lecanemab selectively binds to neutralize and eliminate soluble, toxic amyloid-beta (Aβ) aggregates (protofibrils) that are thought to contribute to the neurodegenerative process in AD. The drug received accelerated approval from the U.S. Food and Drug Administration in January. As Eisai applied for formal approval, the FDA plans to hold an advisory committee on the 9th (US local time) and decide whether to grant approval next month. Unlike 'Aduhelm (ingredient: aducanumab)', which failed commercialization, high expectations are in place for lecanemab’s growth into a blockbuster drug. Clarivate, a global academic information service company, predicted that lecanemab’s sales would reach USD 1.02 billion (KRW 1.32 trillion) in 2027 in its ‘Drugs to Watch’ report that it released earlier this year. Eisai’s application to the MFDS is based on the Clarity AD Phase III and Phase IIb clinical studies that confirmed that lecanemab reduced clinical cognitive decline in early Alzheimer's disease. The Phase III Clarity AD compared lecanemab with placebo in 1.705 patients aged between 50 to 90 with early AD. The primary efficacy endpoint was the change in clinical decline on the global cognitive and functional scale, CDR-SB, compared with placebo at 18 months. Key secondary endpoints were ▲the AD Assessment Scale-cognitive subscale14 (ADAS-cog14), ▲change from baseline at 18 months compared with placebo of treatment in amyloid levels in the brain measured by amyloid positron emission tomography (PET), ▲AD Composite Score (ADCOMS), etc. Change in CDR-SB score with lecanemab -placebo (primary endpoint) (data: Biogen) Results showed that the lecanemab arm recorded a CDR-SB score of 1.21 at 18 months, which is a 27% reduction in clinical decline compared with the 1.66 recorded in the placebo arm. This delay started as early as six months. The drug also achieved statistically significant in its key secondary endpoints compared with placebo. In the amyloid PET subgroup analysis, the lecanemab arm showed a statistically significant reduction in brain amyloid accumulation from 3 months. The ADAS-Cog14 evaluation results showed that cognitive decline was delayed by 26% with lecanemab. ADCOMS results also showed a 24% delay in disease progression at 18 months with lecanemab.