- LOGIN
- MemberShip
- 2026-05-02 14:27:20
- Policy
- Initial appvl rate of drugs subject to prior review varies
- by Lee, Tak-Sun May 18, 2023 05:45am
- As a result of analyzing the approval rate of prior authorization drugs over the past 10 years, the approval rate varied greatly according to the type of drug. However, unlike during the initial review, the review for maintenance therapy showed a high approval rate of 90%. Yong-Kyun Won, Professor of Radiation Oncology at Soonchunhyang University Cheonan Hospital, announced so through a retrospective record analysis study on the prior authorization drugs over the past 10 years (2021-2022).' The study was presented at the 22nd Annual Conference of the Korean Society of Insurance Medicine which was held on the 14th. The prior authorization system was implemented in 2012 to establish clear standards for the use of high-priced drugs and to prevent drug abuse. Soliris, Spinraza, Ultomiris, Strensiq, and Zolgensma, which are rare disease drugs and ultra-high-priced drugs that cost more than KRW 300 million won per year, receive health insurance reimbursement through the system. Crysvita was recently added as a drug that requires prior authorization. According to the study, drugs that were expensive but are essential for the treatment of rare diseases have been able to receive reimbursement through the system, and this pre-deliberation system has been successful, such as in managing the quality of treatment through patient monitoring (maintenance therapy), etc. However, the approval rate was different for each drug. In particular, the prior authorization approval rate for initial administrations ranged from 20% to 100% by product or indication. In comparison, the review approval rate for maintenance therapy exceeded 90%. The varying initial approval rate of prior authorization drugs (retrospective record analysis study on prior authorization drugs (2012~2022)) For example, in the case of patients who seek to use Soliris for aHUS, the initial approval rate was only 21.6%. On the other hand, drugs such as Ultomiris (77.8%) and Strensiq (100%) showed high approval rates. On the other hand, the number of acceptations of objections on the disapproval was low. Only 1 out of 17 objections in 2022 were accepted, and therefore the analysis was that it was a difficult environment for disapproved drugs to receive deliberations again. Professor Won expressed concerns about how the low approval rate may limit access to reimbursement. In addition, for drugs in need of urgent deliberations due to the child's age or disease type, Won explained that the system where institutions need to wait for announcements until the end of the month to see why their application was disapproved and what needs to be supplemented, should be improved as well. Professor Won said, “The prior authorization system is settling as an essential system in securing access to treatment for high-priced drugs that are being continuously introduced to the field. Doctors may feel it is difficult to use a drug if the approval rate is too low. Therefore, it is necessary to review whether the reimbursement standards are too strict and whether it needs revisions.”
- Policy
- Patent protection for 42 yrs for Humira/32 yrs for Keytruda
- by Lee, Hye-Kyung May 16, 2023 09:07pm
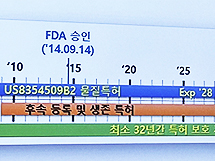
- It was analyzed that Humira maintained its patent protection period for at least 42 years and Keytruda for at least 32 years after filing for material patents with the Evergreening patent strategy. Evergreening refers to extending the term of a patent or extending the term of a patent for more than 20 years in the case of a patent to obtain more exclusive rights during the patent protection period. The types of evergreening strategies are representative of salt compounds, solvates, crystalline forms, optical isomers, dosage forms and pharmacokinetic data, manufacturing methods, and uses. Kim Tae-Kwon, responsible for the Korea Institute of Patent Technology Advancement, held on the last day of the 'Bio Korea 2023' event held on the 12th, 'Strategies for responding to original drugs and late-comer drugs according to the expiration of blockbuster drug patents'. Introducing the patent strategy. Kim Tae-kwon, head of the Korea Institute of Patent Technology Promotion The reason Humira and Keytruda are cited as examples is that the two medicines, excluding vaccines, respectively, ranked first and second in global sales in 2021, with sales of Humira at $20.694 billion and Keytruda at $17.186 billion that year. Humira applied for a substance patent in the United States in 1996, was registered as a patent in 2000, and received FDA approval in 2002. Since then, it has received FDA approval for additional indications such as psoriatic arthritis, ankylosing spondylitis, and Crohn's disease, and the substance patent period expired in 2016. So far, Humira has confirmed 746 patents based on 79 original patent applications. By type, 23 family groups (29.11%) for medicinal use and manufacturing method were distributed the most, followed by other 9 (11.39%), 8 diagnoses (10.13%), 7 formulations (8.66%), and 4 compositions. (5.56%), and 2 material improvements (2.53%). Responsibility Kim said, “Before the substance patent of Humira expired, we continuously extended the patent period with patents for medicinal use, formulation and composition patents, manufacturing method patents, diagnostic patents, automatic administration device patents, and pharmaceutical improvement patents.” It maintained its monopoly." In other words, Humira used an ever-greening patent strategy, such as forming a relatively superior patent barrier in the field of pharmaceutical use and manufacturing methods and forming a patent barrier in the pharmaceutical field as well. As a result, Humira was able to have a protection period of at least 42 years after applying for a substance patent and 35 years after FDA approval. After applying for a material patent in 2008, Keytruda registered a patent in 2013 and received FDA approval in 2014. Based on 67 original applications for patents, a total of 345 patents were confirmed. Looking at the distribution by type, 48 (71.64%) of the medicinal uses showed an overwhelming distribution. Next, 7 drugs (10.45%), 4 double antibodies (5.97%), 3 substances (4.48%), 3 diagnoses (4.48%), and 2 others (2.99%) are shown. Kim said, "There was also a case where the dosage and usage were changed as a barrier patent for the evergreening strategy of Keytruda products." "There was also data that it was to block the development of biosimilars as the patent term expires in 2028." and interpreted. In the case of Keytruda, it used the evergreening patent strategy, and it was analyzed that it had a protection period of at least 32 years after the substance patent and about 26 years after FDA approval. Kim explained, "As a result of the analysis of patent applications by type of biopharmaceutical, material patents must be filed, and based on material patents, applications are filed in the order of pharmaceutical use, composition, formulation, manufacturing method, diagnosis, and material improvement patents." As a result of comparing the evergreening strategy of biopharmaceuticals and synthetic drugs, there are patents for use, manufacturing methods, and formulations after substance patents, but synthetic drugs have many improved patents such as crystal forms, optical isomers, polymorphs, and intermediate patents for chemical formulas. However, as for biopharmaceuticals, there were patents for improving antibody or protein drugs. In addition, substance patents are important, but in the case of manufacturing method patents, it was noted that the distribution of biopharmaceuticals was high and the distribution of synthetic drugs was low. Kim said, "One thing to look carefully at is the expiry of the duration of a biopharmaceutical, but we need to look at changes in the patent for formulation, usage, and dosage in the use patent."
- Policy
- Biopharmaceutical CDMO annual average of ↑31%
- by Lee, Hye-Kyung May 15, 2023 05:41am
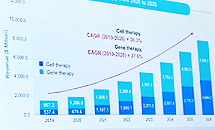
- Kwon Soon-jae, managing director of ENCell, is giving a presentation on the current status of the CDMO market at Bio Korea 2023.Globally, the CDMO market for biopharmaceuticals is growing at an average annual rate of 31%. If this trend continues, the size of the Cell&Gene Therapy CDMO market is expected to reach 10 billion dollars in 2026. Kwon Soon-jae, managing director of ENCell, announced this at the 'CDMO Partnership for Acceleration of Biopharmaceutical Development session held at the 'Bio Korea 2023' event held on the 10th. Director Kwon explained, "Cell & Gene Therapy is expected to grow 5.5 times in 2026 compared to 2019, and gene therapy is expected to grow 8.7 times." He explained, "If you look at the high CAGR from 2019 to 2026, it will account for 36.3% and 27.6%, respectively." The demand for CMOs and CDMOs has increased due to the COVID-19 pandemic, and Director Kwon said, "Small companies use CMOs and CDMOs to reduce costs and time, while large companies use CMOs and CDMOs to reduce marketing and R&D costs." It plays a part," he said. However, in the case of domestic CGT treatment, the manufacturing technology is complicated and the number of platforms is not large, so it was inevitable to create a GMP facility with an 'in-house' concept rather than CDMO service, and have many in-house processes and services. Director Kwon said, "However, as the requirements of the Ministry of Food and Drug Safety become stricter, infrastructure, raw materials, facility costs, labor costs rise, and technology becomes more complex, outsourcing instead of in-house is becoming a trend." Looking at the domestic CGT CDMO market, Lonza, Samsung Bio, SK, CJ, Lotte, and Medipost have announced their entry into the CMO/CDMO business following Thermoficer in 2017. Director Kwon said, "Most of the 30 CGT companies in Korea are major companies, and 80% of them are trying to develop AAC, adenovirus, CAR-T, etc., and only 30% of them are investing more than 2 million dollars." "If you look at the CGT market alone, it's still the first step, the introductory stage," he said. Director Kwon said that the present, when the first step of CDMO in the CGT market was taken, is an important point in determining the future. Director Kwon said, "More than 100 companies worldwide have entered the CDMO cell gene therapy market, and price, location, and regulations are challenges to be resolved." It looks like I'll have to give it a try," he said.
- Policy
- Youkyung Oh was appointed as the first chairman of APFRAS
- by Lee, Hye-Kyung May 12, 2023 05:44am
- On the 10th and 11th, the Ministry of Food and Drug Safety (Minister Oh Yookyung) held the 1st Asia-Pacific Food Regulatory Authority Heads Consultative Meeting (APFRAS 2023), where 7 countries came together to harmonize global food regulations in the Asia-Pacific region and strengthen cooperation. said to have collected. APFRAS (Asia-Pacific Food Regulatory Authority Summit) is participated by New Zealand, Vietnam, Singapore, China, Philippines, Korea, and Australia. On the 11th, Korea was elected as the first chair of APFRAS at a meeting of heads of food regulatory agencies from seven countries, and Minister Oh You-keong of the Ministry of Food and Drug Safety was appointed as chair. As the chair, Korea established a secretariat, operated a working group, and communicated among member countries for three years. do The member countries adopted the Operational Regulations (TOR) following the establishment of APFRAS, and also resolved implementation tasks for the operation of the working group and achievement of strategic goals. In the future, the APFRAS working group will analyze the food regulatory environment in the Asia-Pacific region and discuss in-depth discussions on the digitalization of food safety management and carbon neutrality in the food sector. In addition, for food safety, we agreed to rapidly analyze new global issues and respond cooperatively to changes in the international food environment, and urge the strengthening of the cooperative system to create a safe food distribution environment in the Asia-Pacific region and solve common tasks. The Declaration was adopted and signed by all seven member states. It was agreed to hold the APFRAS meeting once a year to continue the close cooperation system among member countries, and the second APFRAS meeting in 2024 is scheduled to be held in Seoul, Korea, which was elected as the chair country. Minister of Ministry of Food and Drug Safety Oh Youkyung discussed with Lim Kok Thai, head of the Food and Drug Administration of Singapore, to conclude a food safety cooperation agreement (MOU) to draw common interests between the two countries, such as standards for new food raw materials, and to strengthen cooperation between the two organizations. Director Oh said, "With the launch of APFRAS, the world's first head of a food regulatory agency gathered in one place to discuss various issues related to global food safety and achieve meaningful results by agreeing on strengthening capabilities among regulatory agencies." Director Oh said, "As I was elected as the APFRAS chairman, I will do my best so that Korea can play a leading role in quickly identifying new food safety issues and changes and raising the level of food safety in member countries. I will try,” he said. The Ministry of Food and Drug Safety will continue to lead international cooperation and regulatory harmonization for food safety and continue discussions to resolve non-tariff barriers. .
- Policy
- HIRA plans to report on the re-evaluation
- by Lee, Tak-Sun May 12, 2023 05:44am
- It is known that The HIRA will come up with a plan to improve the re-evaluation of drug benefits and report it to the Health Insurance Policy Review Committee of the Ministry of Health and Welfare, which will be held this month. This improvement plan is based on the 'rationalization plan for re-evaluation of drug benefit adequacy', which ended in March. According to the industry on the 11th, The HIRA is conducting internal procedures to disclose the results of the 'rationalization plan for re-evaluation of drug benefit adequacy', which was conducted as an external service research. Based on this, improvement plans are said to be reported this month. Along with the health report, the results of the research service will also be made public. It is known that the service research conducted by KIHASA (Research Director: Dr. Sylvia Park) contains the direction of re-evaluation and rational operation plan. Accordingly, the number of registered drug products and claims for the past 10 years were analyzed, and the claims for first-listed ingredients after 2007 were also analyzed. The pharmaceutical industry is also paying attention to this improvement plan as it is known that it will affect the decision to be re-evaluated in the future. In particular, the ingredients subject to a re-evaluation of benefit adequacy in 2024, which have not yet been announced, are also being selected based on this, so they are keenly aware. An official in the pharmaceutical industry said, "We are keeping an eye on the related contents because the results of the research service show the direction of the selection of ingredients subject to a re-evaluation of benefits in the future." It is known that HIRA plans to proceed with the process of selecting target ingredients in 2024. An official from HIRA explained, "We plan to start selecting target ingredients in 2024 through a subcommittee." Meanwhile, the re-evaluation of benefit adequacy will be conducted in 2020 on drugs with low clinical usefulness to optimize pharmaceutical expenditure. In 2020, Choline alfoscerate was re-evaluated, and in 2021, ingredients mixed with healthy functional foods were re-evaluated. Last year and this year, 6 ingredients were selected in 2022 and 8 ingredients in 2023 were selected based on the old ingredients, etc., and the review is underway, such as determining the conditional temporary benefit for streptokinase and streptodornase ingredients. This year, drugs such as hyaluronic acid eye drops are on the re-evaluation judgment table for the adequacy of reimbursement.
- Policy
- Triple-negative breast cancer tx Trodelbi approved in Korea
- by Lee, Hye-Kyung May 11, 2023 05:50am
- Meditip's Trodelbi (Sacituzumabgovitecan), an orphan drug triple-negative breast cancer treatment, has received domestic product approval. The Ministry of Food and Drug Safety (Minister Oh Yoo-kyung) announced on the 9th that it had approved the approval of Trodelbi to be used in breast cancer patients who lack estrogen receptor (ER), progesterone receptor (PR), and epidermal growth factor receptor 2 (HER2). Trodelbi is an antibody-drug conjugate that targets Trop-2 protein, frequently found on the surface of breast cancer cells, and provides a new treatment opportunity for patients with advanced or metastatic triple-negative breast cancer. Trop-2 is overexpressed on the surface of various cancer cells, including triple-negative breast cancer. Trodelbi is indicated for treating adult patients with unresectable locally advanced or metastatic triple-negative breast cancer who have received at least two prior systemic treatments, at least one of which has been treated for metastatic disease. Trodelbi induces the death of cancer cells by releasing drugs (SN-38, SN-38 glucuronide) that inhibit cell division within the cell, while the antibody (Sacituzumab) binds to Trop-2 expressed on the cell surface and moves into the cell. do. The Ministry of Food and Drug Safety said, “We will continue to do our best to promptly supply treatments whose safety and effectiveness have been sufficiently confirmed based on regulatory science.”

- Policy
- ‘Pay more policy attention to advanced heart failures'
- by Hwang, byoung-woo May 11, 2023 05:49am
- [Interview with Medical Societies] Soo-Yong Lee, Administrative Secretary of the Insurance Committee at KSF Asks authorities to increase benefits for patients at high risk of health failure who have fewer treatment alternatives According to the ‘2020 Heart Failure Fact Sheet’ that was released by the Korean Society of Heart Failure (KSHF), Korea’s prevalence of heart failure in Korea had increased threefold in 16 years from 0.77% of the total population in 2002 to 2.24% in 2018 to exceed 1 million patients. Although drug options that can intervene in the early stages of heart failure have been increasing, options are still limited for severely ill patients with prior hospitalization experience. Therefore, Soo Yong Lee, Professor of Cardiology at Pusan National University Yangsan Hospital (Administrative Secretary, Insurance Committee, KSF) believes that appropriate policy intervention is needed in terms of patient benefits and insurance finance. Soo-Yong Lee, Assistant Administrator of the Insurance Committee at KSF#In particular, Professor Lee stressed how heart failure has a lower survival rate than most cancers. “The overall survival period of patients with heart failure is 2.6 years for first hospitalizations, 1.8 years for second hospitalizations, and 1.5 years for third hospitalizations. This means that the number of hospitalizations is proportional to the mortality rate of the patients, and 1-2 out of 4-5 patients are re-hospitalized within a month in practice.” Lee further explained that hospital readmissions also impose further financial burdens on the patients. The total medical expense paid by patients with heart failure who have experienced at least 1 hospitalization is around KRW 8-9 million per year, and the burden increases further if the patient’s condition requires the use of an intensive care unit or equipment for dialysis or ECMO. In fact, according to the 2017-2021 health insurance treatment Rep. Sun-woo Kang, member of the National Assembly's Health and Welfare Committee, received from the National Health Insurance Service, the number of patients treated for heart failure increased by 7.1% (158,916 in 2021) every year, increasing the treatment expense as well (an average of 15.6% in 5 years). The heart failure treatment paradigm has been changing with recent studies being conducted on reducing the mortality rate in patients with chronic, therefore, stable heart failure and the introduction of ARNi drugs. Lee said, “Recent studies have focused on how much the condition improves when drugs are used in acute patients after treatment and when drugs are used immediately after stabilization. With the release of SGLT2is and ARNis, the current trend is leaning towards the early use of such treatments” Re-hospitalization of patients despite the availability of early treatment options remains a concern..."Need to improve the treatment environment" However, despite the development of early treatments, the number of readmitted patients has increased constantly due to various factors including the lack of patients' compliance. One treatment that can be considered for use in this situation is vericiguat (product name Verquvo), and the KSF has been highly recommending it with a Class Ⅱa recommendation for preemptive use when a patient’s heart failure worsens even after ample standard therapy. The VICTORIA trial that became the basis of Verquvo’s approval drew attention because it enrolled patients who have recent hospitalization history and have been hospitalized at least once. Compared to most studies of other heart failure drugs that are conducted on chronic patients with good symptom control and low readmission rates, Verquvo’s patient group fundamentally has a higher mortality rate than other studies. Lee said, “In the VICTORIA study, 66.9% of patients were hospitalized for heart failure within 3 months, and 85.7% were HFrEF patients with a left ventricular ejection fraction of 40% or less. The study itself was a brave attempt as most of them were in a very bad condition, to the extent that no drugs would have been effective for them.” Study results showed that Verquvo reduced the risk of death from cardiovascular disease or first hospitalization due to heart failure by 10%, and achieved a 4.2% reduction in annualized absolute risk. Regarding the results, Lee explained that “Patients in the high-risk group used to have a poor prognosis. They were prescribed dobutamine before and are discharged if they seem better, and had to repeat hospitalization due to cardiac arrest until death or await heart transplantations. The study showed that its NNT was 24, which means that 1 out of 24 patients could be discharged because their symptoms improve after using the treatment, which is a very good figure and the best level achieved among heart failure drugs.” He added, “The drug holds great clinical significance as it gives high-risk patients the opportunity to leave the hospital. "In my practice of treating many patients with end-stage heart failure, Verquvo is definitely a welcome rain in the drought.” Emphasis on its benefit in patients at high risk of heart failure...will reimbursement discussions for Verquvo make progress? According to industry sources, Verquvo’s reimbursement has passed review by the Health Insurance Review and Assessment Service’s Drug Reimbursement Standard Subcommittee and is awaiting to be deliberated by the Drug Reimbursement Evaluation Committee. To add its support, the KSHF has also conveyed its opinion regarding the expansion of Verquvo’s reimbursement standards as its role in the field is clear. Based on the VICTORIA trial, the KSHF expects that 10,000 to 15,000 patients can be treated with Verquvo every year. In particular, Lee judged that when the drug is administered to the high-risk group, this may reduce the need for a heart transplant or hospitalization in an intensive care unit, which can also provide benefits in terms of cost. He said, “The biggest feature of Verquvo is that it has confirmed its effectiveness in severely ill patients. When considering how patients with the LVAD indication incur KRW 150 million to KRW 250 million as expenses every time they use LVAD, if the drug can reduce the frequency of hospitalization or death, reimbursement would also be reasonable in terms of saving insurance finances.” However, Lee expressed concern over how the application of excessively restrictive reimbursement standards may act as a barrier to its use for patients even if the drug is positively considered for reimbursement in the future. In the VICTORIA clinical trial, about 60% of the patients received the three-drug therapy that included RAAS inhibitors. Patients who experienced worsening conditions despite being administered standard therapy according to the patient’s clinical condition were also allowed to use Verquvo in the trial, and therefore this indication may also be reflected in its reimbursement standards in the future. However, as standard therapy treatments are used in primary medical institutions in the early stages of heart failure, a barrier may arise for patients where they may not be eligible to use Verquvo even after being transferred to a general hospital or a tertiary hospital due to set reimbursement standards. Lee said, “I think Verquvo is a necessary drug for patients in the advanced stage, such as those who have used intravenous diuretics or have been hospitalized for heart failure. As a clear patient population exists for the drug, its reimbursement standards should be set promptly in consideration of the urgent need and allow its use in patients who experience worsening heart failure events after standard treatment.”

- Policy
- Rapidly changing breast cancer treatment
- by Choi, sun May 11, 2023 05:49am
- The Korean Breast Cancer Society revised the treatment recommendation on the 27th. Combination drug treatment using a combination of an antibody and an anticancer drug has emerged as a hot issue, and as the new anticancer drug Enhertu for HER2-positive metastatic breast cancer received domestic approval in September last year, the reflection of this is emerging as a concern. In the case of new drugs, society reflects them if there is evidence, while also preparing new recommendations for rare cases that have been neglected. It means that it presented an 'answer' based on expert consensus in areas where large-scale randomized clinical studies were lacking due to the small number of patients, such as male breast cancer, osteoporosis treatment in breast cancer patients, and familial breast cancer, which depended on individual judgment of medical staff. Given that the clinical field of breast cancer is rapidly changing due to the emergence of various new drugs and treatments, society adheres to preparing revisions every two years. Even with a 'short cycle' of 2 years, it contains a lot of changes. We met Ae-Ri Han (Department of Breast Surgery, Yonsei Wonju University) and In-Hye Park (Department of Oncology, Korea University Guro Hospital), chair of the Breast Cancer Society, to hear about major changes. Usually, guidelines and recommendations are based on data. After the evidence is accumulated and verified over time, it goes through the usual procedure reflected in the guidelines. The problem is in the case of rare cancers, where it is difficult to accumulate data despite the passage of time. The need for a minimum 'guidance' that relied entirely on the judgment of medical staff has been a demand in the clinical field. (From left) Han Ae-ri, chair of the breast cancer society, and Park In-hye, chair of the academic committee Chairman Han expressed the biggest change in this recommendation as 'interest in the minority. "Because recommendations are not standard medical guidelines, they do not mean that they must be done as they are," she said. Usually, for rare cases, foreign studies are referred to. It was not easy to find high-quality research data abroad for the rare cases included in this guideline. Chairman Han said, "The most reliable data is a randomized clinical trial involving a long period of time and a large number of patients, but the cases mentioned above have physical limitations in conducting such clinical trials. This is the same situation in Korea as well as abroad." Explained. The society decided to support smooth use through recommendations on the use of Enhertu, which is on the verge of reimbursement. Enhertu drew attention last year with a national consent petition urging 50,000 people to request rapid approval. Even after the domestic approval in September of last year, as 50,000 people urged public consent for health insurance, it emerged as a topic of interest in the breast cancer academic community. Park In-hye, chairman of the Academic Committee (Korea University Guro Hospital), said, “Enhertu’s insurance review has already been completed and some adjustments remain, so the review will begin again in May soon.” Since locals use it a lot, I think a similar level of decision will come out." She said, "I think that insurance benefits will be available to patients after the review in May in Korea." She said, “Especially, as the treatment indications for Enhertu are getting wider, the number of patients who can be treated with Enhertu is expected to gradually increase.” Chairman Han Ae-ri said, "Because the level of evidence must be high, it is difficult to unconditionally reflect in the recommendation that a new drug has been released, but all cases that meet the criteria such as Enhertu are reflected in this guideline." I thought it was, so I didn't reflect it," she explained. “The National Comprehensive Cancer Network (NCCN) has recommended Ribociclib as a first-class among CDK 4/6 inhibitors,” she said. “In Korea, Palbociclib was first launched in 2016 and is a generic drug. Abemaciclib and Ribociclib are competing, but experts are also divided on whether to switch to another drug if they are currently taking Palbociclib.” Although not enough data has been accumulated to change the recommendation, it was not easy to make a decision because the recommendation level for late-comer drugs is being raised overseas. In particular, it was also pointed out that if the prognosis worsens after first administering Ribociclib, there is no other drug that can be used. Chairman Han said, “There was an opinion that existing drugs should be used first and new drugs should be used as a last resort in preparation for a worsening prognosis, but in the end, there were more opinions that good drugs should be used from the beginning.” I also gave a lecture about using good medicine first from the beginning.” "Currently, the market is changing due to competition in generics such as Ribociclib, and the recommended level is also changing, so it is true that there is confusion in the clinical field," said Han. Chairman Han added, "If there is an effective treatment, I think it is the mission of the society to reflect and recommend it."
- Policy
- A benefit study of the Gardasil 9 NIP is also forthcoming
- by Lee, Jeong-Hwan May 10, 2023 11:17pm
- The KDCA is ordering an additional policy research service to apply the National Vaccination Support Project (NIP) of the 'HPV 9-valent vaccine' to female adolescents and male adolescents over 12 years of age. As the result of the HPV 9 commissioned by KDCA to NECA earlier was found to be low in cost-effectiveness in vaccine research, it is in effect a follow-up study. In addition, the KDCA plans to conduct a cost-effectiveness study when applying NIP to the shingles vaccine. On the 8th, KDCA Medical Safety and Prevention Director Lim Eul-ki made this statement at a meeting with the Professional Reporters Association. Director Lim Eul-ki explained, "The additional study on the cost-effectiveness of the male HPV 9 vaccine will be conducted as quickly as possible and an order will be placed in May." The policy of free vaccination of Gardasil 9, a vaccine against HPV 9, to female and male adolescents was a pledge of President Seok-Yeol Yoon during the presidential election. To this end, the KDCA commissioned NECA to conduct a cost-effectiveness study, but it was concluded that it was not cost-effective in all analysis scenarios. Specifically, the research team analyzed the economic effects of three scenarios: ▲ 12-year-old girl 9-valent conversion, ▲ 12-year-old male and female 9-valent vaccination, ▲ and 12-year-old male HPV vaccine NIP subject to NIP However, as a result, the cost-benefit ratio was not significant in all scenarios. Accordingly, KDCA decided to conduct a cost-benefit analysis again through additional research. The subject of the follow-up study was decided to be kept private for the time being to maintain research fairness. Regarding the background of the additional research, Director Lim Eul-ki explained, "There were many experts' opinions that the research design of the research service conducted by NECA was carried out excessively conservatively." Director Lim explained, "For example, we underestimated the diseases that occur in men during the HPV vaccine effect." Director Lim said, "There was an expert opinion that the effect on side diseases such as head and neck cancer was also underestimated. In follow-up studies, we plan to actively reflect this aspect." Director Lim added, “In particular, it should have been studied compared to female non-vaccinated people, but there was also an evaluation that the sensitivity was lowered because of the study compared to female non-vaccinated people.” Director Lim added, "In fact, research services have been conducted overseas as well, and additional research is being ordered in Korea under the same conditions." The KDCA will also embark on a NIP cost-benefit study of the shingles vaccine. The research will also be ordered in May. Director Lim said, "It would be nice if NIP was implemented for the shingles vaccine, but it was difficult to proceed due to budget limitations. This research service will include the recently released Shingrix vaccine." Lim said, "Unlike HPV vaccine research, the period required for research on shingles is planned to be about one year with time to spare."
- Policy
- Prior HIRA approval is required to administer Crysvita
- by Lee, Tak-Sun May 10, 2023 06:00am
- Prior approval from the Health Insurance Review and Assessment Service will be required to administer the pediatric rickets treatment Crysvita Inj which is set to be reimbursed from May this year. Accordingly, HIRA has prepared the specifics for Crysvita’s prior approval and applied it from the 3rd of this month. According to the specific criteria, medical institutions that seek to administer Crysvita’s to children with X-linked hypophosphatemia (XLH), a rare inherited form of rickets, must apply for prior approval to HIRA. The deliberation will be conducted by the Crysvita subcommittee under the Healthcare Review and Assessment Committee (HCRAC). The subcommittee will convene on the third Wednesday of each month and will be deliberating applications submitted until 14 days before the date of the meeting. Medical institutions that receive HIRA’s Prior approval must administer Crysvita within 60 days of being notified of the deliberation results. If the institution seeks to administer it after 60 days, it must reapply for prior approval. Medical institutions that have been approved in advance and administered Crysvita can file claims for health insurance benefits. The institutions are also required to submit monitoring data after administering Crysvita. The medical institution in charge is required to submit a monitoring report every 12 months after initiating treatment before administering the maintenance dose, which is again subject to the subcommittee’s review. The subcommittee can reverse its approval if the institution approved to receive the health insurance benefits is found to have falsely filed related data in the applications and monitoring reports or submitted false data. However, a medical institution that has been notified of the subcommittee’s decision to cancel or withdraw the approval of health insurance benefits may file an objection within 90 days. With the addition of Crysvita Inj., a total of 9 items now require prior approval before being insurance benefits: ▲Immune Tolerance Induction; ▲Soliris‧Ultomiris Inj. ▲Strensiq Inj. ▲Spinraza‧Zolgensma Inj.; ▲ Autologous Stem Cell Transplantation; ▲Implantable Cardioverter Defibrillator & Cardiac Resynchronization; ▲Ventricular Assist Device therapy; ▲Clinical studies; and ▲Crysvita Inj. However, in the case of autologous stem cell transplantation, institutions can opt to receive a review after administration without undergoing prior approval procedures.